
Abstract
As arguably the simplest free-living animals, placozoans may represent a primitive metazoan form, yet their biology is poorly understood. Here we report the sequencing and analysis of the ∼98 million base pair nuclear genome of the placozoan Trichoplax adhaerens. Whole-genome phylogenetic analysis suggests that placozoans belong to a ‘eumetazoan’ clade that includes cnidarians and bilaterians, with sponges as the earliest diverging animals. The compact genome shows conserved gene content, gene structure and synteny in relation to the human and other complex eumetazoan genomes. Despite the apparent cellular and organismal simplicity of Trichoplax, its genome encodes a rich array of transcription factor and signalling pathway genes that are typically associated with diverse cell types and developmental processes in eumetazoans, motivating further searches for cryptic cellular complexity and/or as yet unobserved life history stages.
Similar content being viewed by others


Main
Placozoans (literally, ‘flat animals’) are small (1–2 mm), disc-shaped creatures that were initially discovered1 on the walls of a saltwater aquarium in the late 1800s. These unusual animals were largely neglected until they were rediscovered2 in the 1970s and were subsequently found throughout tropical and subtropical oceans in nearshore habitats, particularly mangrove communities3,4. Placozoans are readily collected in the wild and can be maintained in the laboratory on diverse food sources. Although placozoans found in diverse locations are morphologically indistinguishable, they show surprising diversity at the DNA level, suggesting that cryptic species may exist5,6,7. The only named species in the phylum is Trichoplax adhaerens F. E. Schulze1.
Trichoplax appears as a flat disc of cells consisting of two epithelial layers, which sandwich a layer of multinucleate fibre cells (Fig. 1a). Only four cell types have been described previously8,9 (Fig. 1b); nerves, sensory cells and muscle cells are apparently absent. To feed, Trichoplax climbs atop its food using the bottom surface as a temporary extraorganismal gastric cavity; digestion is both extracellular and phagocytic10,11. When not feeding, the animals move by cilia on the bottom surface and by the fibre cell layer10. Placozoans have no evident body axes other than top versus bottom and periphery versus interior; they show no regular directionality in their movement, although both positive and negative phototaxes have been observed (K. von der Chevallerie, T. Bergmann and B. Schierwater, unpublished observations).

a, Trichoplax in laboratory culture; scale bar, 200 μm. b, Schematic rendering of a transverse section through Trichoplax. B, bacterium in endoplasmic cisterna; FC, contractile fibre cell; GC, gland cell; LE, lower epithelium; MC, mitochondrial complex; SS, shiny sphere; UE, upper epithelium (taken with permission from ref. 45). c–e, Trichoplax progressing through asexual reproduction by fission; scale bars, 200 μm. f, A cleaving Trichoplax ‘embryo’ at the 4-cell stage is shown; scale bar, 20 μm. g, A cleaving Trichoplax ‘embryo’ at the 16-cell stage is shown; scale bar, 20 μm.
In culture, Trichoplax reproduces by fission, whereby two (sometimes three) parts of the animal move away from each other until their connection is ruptured (Fig. 1c–e). Sexual reproduction has not been observed in culture but putative oocyte formation in degenerating animals is routinely seen12. These large cells have been observed to undergo cleavage (Fig. 1f, g) up to a 256-cell stage before degenerating (M. Eitel and B. Schierwater, unpublished observations). Although sperm have been described previously13, this has not been seen by other investigators. Population genetic analyses, however, demonstrate allelic variation and evidence for genetic recombination in animals in the wild that is consistent with sex14.
The phylogenetic relationship of placozoans to other metazoans remains controversial. Whereas early studies on the basis of a small number of genes suggested that placozoans could be secondarily simplified cnidarians15, other analyses refuted this position16,17. Small subunit ribosomal RNA analysis suggested placozoans as eumetazoans, either as a sister group to bilaterians16 or as the earliest eumetazoan branch18 (although addition of the large subunit rRNA sequences reduced the resolution of the phylogenetic tree). Analyses of the complete mitochondrial genomes of Trichoplax adhaerens19 and other placozoans20, however, led to the proposal that Placozoa could be the earliest branching basal metazoan phylum (but see ref. 21).
Here we report the draft nuclear genome sequence of Trichoplax adhaerens and use it to begin to address the nature of placozoans. Our phylogenetic analysis supports the identification of placozoans as a basal eumetazoan lineage that diverged before the separation of cnidarians and bilaterians but after the divergence of demosponges from other animals. The compact genome shows remarkable complexity, including conserved gene content, gene structure and synteny relative to human and other eumetazoan genomes. Despite the absence of any known developmental program and only a modest number of cell types, the Trichoplax genome encodes a rich array of transcription factors and signalling genes that are typically associated with embryogenesis and cell fate specification in eumetazoans, as well as other genes that are consistent with cryptic patterning of cells, unobserved life history stages and/or complex execution of biological processes such as fission and embryonic development in these enigmatic creatures.
The Trichoplax genome
We produced a high-quality draft sequence of the ∼98 million base pair (megabases, Mb) Trichoplax genome using whole-genome shotgun methods22 with ∼8-fold redundant sequence coverage. Because there are at present no genetic or physical maps of Trichoplax, we could not reconstruct entire chromosomes, but the completeness of the draft assembly (98% of the 14,571 expressed sequence tags (ESTs) align) and its long-range linkage (19 scaffolds longer than 1 Mb represent 80% of the assembly) make it an excellent substrate for annotation and comparative analysis (Supplementary Information).
As expected from genomic sequences derived from an asexually reproducing laboratory culture of diploid animals, only two alleles are observed at each locus. The single nucleotide polymorphism frequency is 1% and is distributed as expected for two haplotypes selected from a panmictic sexual population (Supplementary Information). We observed 35 extended regions of unusually low polymorphism (<0.25% over more than 40 kb), indicating recently shared ancestry of the two haplotypes by inbreeding or gene conversion and/or the influence of selective sweeps. Sampling of more than two haplotypes is required to distinguish between these two possibilities.
Trichoplax gene complement and conserved gene structures
We estimate that the Trichoplax genome contains 11,514 protein coding genes, on the basis of a combination of homology-based and ab initio methods (Supplementary Information). Nearly 87% of these predicted genes have detectable similarity to proteins known from other animals, and most (83%) of the ∼7,800 gene families that are conserved between the sea anemone and bilaterians23 have homologues in Trichoplax as detected by BLAST. Trichoplax genes have an intron density (7.6 per kb) comparable to that found in vertebrates (8.5 per kb) and the starlet sea anemone (6.7 per kb)23.
Analysis of the exon–intron structure of orthologous genes demonstrates a high degree of conservation in Trichoplax relative to other eumetazoans, extending the antiquity of many animal introns (Supplementary Information)23. For example, in conserved regions, 82% of human introns have orthologous counterparts with the same position and phase in Trichoplax. The retention of ancient introns in Trichoplax is in contrast to other animals with small genomes that show extensive intron loss (for example, fruitfly, soil nematode and sea squirts) that presumably accompanied their reduction in genome size23.
Relationship of Trichoplax to other animals
We reassessed the phylogenetic position of placozoans relative to other metazoans using Bayesian, maximum likelihood, and parsimony analyses of a concatenation of 104 slowly evolving single-copy nuclear genes (6,783 aligned amino acid positions) drawn from nine diverse fully sequenced genomes (Fig. 2 and Supplementary Information). With 100% Bayesian support and 92% likelihood bootstrap support, placozoans are found to be a sister group to the other eumetazoans (as represented by two cnidarians and a sampling of diverse bilaterians), with demosponge sequences diverging before the Trichoplax–cnidarian–bilaterian clade. This topology is further supported by parsimony analysis (albeit with weaker support). There is no support for Trichoplax as a derived or basal cnidarian or bilaterian, and these hypotheses are rejected by statistical phylogenetic tests (see Supplementary Information). Although there is strong likelihood bootstrap and Bayesian support for the topology in Fig. 2, our analysis can only reject the placement of Trichoplax basal to other animals at the P = 0.07 level.

Bayesian phylogeny of metazoans places Trichoplax as the sister group to cnidarians and bilaterians. Maximum parsimony applied to the same alignment results in a single tree with the same topology shown here. Posterior probabilities are reported above each branch; likelihood bootstrap support values are reported below.
Although our result disagrees with results from mitochondrial trees19,20,24, these other analyses are complicated by the long branch lengths (that is, unusually high amounts of amino acid divergence) found in bilaterian mitochondrial peptides relative to their basal metazoan orthologues24,25. Figure 2 shows that peptides encoded by the nuclear genome have no notable differences in amino acid substitution levels between basal metazoans and bilaterians, suggesting that our proposed phylogeny on the basis of nuclear genes is less susceptible to long-branch attraction artefacts.
Conserved synteny with other eumetazoans
Although the placozoan lineage diverged from that of other animal phyla in the Precambrian, we find evidence for limited conserved local gene order as well as substantial blocks of longer-range conserved linkage (synteny) in the Trichoplax genome relative to the larger vertebrate and the starlet sea anemone genomes (Supplementary Table 8.1). This is in sharp contrast to the relatively small genomes of flies and nematodes, which show no such conservation. Quantitative analyses of gene neighbourhoods in Trichoplax, human and Nematostella genomes show that the Trichoplax genome has the lowest amount of local rearrangement relative to the common placozoan–cnidarian–bilaterian ancestor (Supplementary Information).
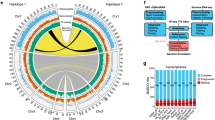
The Trichoplax genome also shows larger blocks of conserved synteny (that is, conserved linkage without requiring colinearity26) relative to the human genome. Each of the 21 longest gene-rich Trichoplax scaffolds contains segments with a significant concentration of orthologues on one or more human chromosome segments (Supplementary Information). These segments are clearly visible in a dotplot of the Trichoplax scaffolds versus the 17 ancestral chordate linkage groups27 (Fig. 3), which shows that many of these linkages have been preserved in the Trichoplax genome, and that most of the chordate linkage groups date back to the placozoan–vertebrate last common ancestor. Neither flies nor nematodes show such conservation. For example, chordate linkage group 10 (comprising portions of human chromosomes 1q, 6p and 9q) appears in its entirety as a relatively compact 3.2 Mb segment of Trichoplax scaffold 2, with substantial gene-order changes (Fig. 3 and Supplementary Fig. 8.1). The observation of blocks of conserved synteny is consistent with a relatively low rate of local rearrangement in Trichoplax.

Blue dots represent orthologues in the 21 most gene-rich Trichoplax scaffolds and the human genome. Human chromosomal segments have been grouped by ancestral chordate chromosome27 (Supplementary Information). Horizontal lines divide groups of human segments descended from each of the 17 ancestral chordate chromosomes; vertical lines divide Trichoplax scaffolds; alternating bars outside the dotplot represent individual human segments (vertical) or Trichoplax scaffolds (horizontal). Red bars and dotted lines highlight the genomic regions compared in Supplementary Fig. 8.1.
Putative developmental transcription factors
With only four (or possibly five28) morphologically identifiable somatic cell types, one might naively expect Trichoplax to possess few transcription factors associated with the complex regulation of cell fate, patterning and differentiation that are found in other eumetazoans (Table 1). Nevertheless, targeted studies of homeoboxes in Trichoplax have identified a modest complement of these essential eumetazoan patterning genes, including paired box genes29 and members of the ANTP class30. Two of these (Trox-2 and Not) are expressed around the rim of the animal, defining the only known molecular patterning of its body plan28,31.
Trichoplax contains a rich repertoire of transcription factors (Table 1 and Supplementary Information) commonly associated with patterning and regionalization during eumetazoan development, including further homeobox-containing genes from the ANTP, Paired (PRD), POU and SIX subfamilies32. Trichoplax also has members of many subfamilies of the animal-specific Sox (Sry-related HMG-box) family involved in the regulation of embryonic development, the T-box family including brachyury (the expression of which defines the blastopore in eumetazoan gastrulation33), and the opisthokont (animal and fungi)-specific Fox (forkhead/winged-helix) family.
Transcription factors that regulate cell type specification and differentiation in bilaterians are also abundant in Trichoplax, including multiple LIM-homeobox genes typically associated with subtype specification in neurons, multiple basic helix–loop–helix family genes associated with neural and muscle cell fates, a (linked) pair of POU–homeobox family genes implicated in neuroendocrine development, and a pair of GATA-family zinc-finger transcription factors that participate in the specification of endodermal, cardiac and blood cell fates. Thus, the Trichoplax genome encodes a variety of nominally cell-type-specific markers despite having only a few recognizable cell types, suggesting massive redundancy of function, alternative functions not directly analogous to those in other animals, or cryptic cellular or developmental complexity.
Putative developmental signalling pathways
Trichoplax lacks consistent directionality in its movements, and possesses upper–lower and centre–rim axes that show no evident homology to bilaterian anteroposterior or dorsoventral axes. Yet, components of a complete Wnt/β-catenin signalling pathway—used for axial patterning in bilaterians and cnidarians34 and in demosponge larvae35—are present in Trichoplax (Fig. 4a). All essential components of the TGF-β signalling pathway are also present in the Trichoplax genome (Fig. 4b). In bilaterians, TGF-β signalling (mediated by BMP, one class of TGF-β superfamily ligands) is responsible for the establishment of the embryonic dorsoventral axis during bilaterian development with a similar axis-defining role proposed in cnidarian36 and demosponge larvae35.

a–d, Known signalling and biological processes in bilaterians are shown in schematic form, with the colours of the protein names indicating their presence (green) or absence (red) in the Trichoplax genome. Wnt/β-catenin signalling (a), TGF-β signalling (b), synapse formation and conduction of nerve impulse (c) and cell adhesion and extracellular matrix components (d) are shown.
We did not find evidence for a functioning hedgehog pathway, because there is no evident hedgehog ligand, patched or smoothened receptors, or Gli-like transcription factor. Components of other signalling pathways such as the Notch and JAK/STAT pathways are present, but these pathways seem to be incomplete in that they lack molecular components critical to signal transduction (for example, a Notch-like gene with a true Notch domain in the Notch pathway, or a Janus kinase in the JAK/STAT pathway; Supplementary Table 9.1). Four nuclear receptor family transcription factors are present, suggesting that signalling by lipid-soluble ligands occurs (Table 1). Elements of the animal stress response NF-κB pathway are found in Trichoplax, along with a nearly complete set of orthologues of genes typically involved in eumetazoan apoptosis (except for TNFR and Fasreceptor; Supplementary Table 9.1).
The absence of components of some animal signalling pathways in Trichoplax relative to their completeness inferred in the cnidarian–bilaterian ancestor23 suggests that Trichoplax branched off from an ancestor that either did not possess all animal signalling pathways or that these genes were lost in the placozoan lineage. This latter interpretation is consistent with the presence of some of these ‘missing’ components (hedgling, EGFR, notch) in sponges37,38,39.
Elements associated with neuroendocrine function
Although Trichoplax has no nervous system, it has behavioural responses to environmental stimuli, and sensitivity to the neuropeptide RFamide has been reported40. In the Trichoplax genome, we find various ion channels that are implicated in neural signalling in animals. For example, different members of the Kv family of voltage-dependent potassium channel α-subunits (the electrically active shaker and shaw and the electrically inactive Kv9) and β-subunits (KCNAB) are present in the Trichoplax genome, along with inward rectifier potassium channels and homologues of voltage-gated sodium channels and voltage-gated L-type calcium channel α1 subunits and their regulatory β-subunit (Fig. 4c).
Components of neurotransmitter biosynthesis and vesicle transport systems, as well as a putative neuroendocrine-like secretory apparatus, are also found in the genome (Fig. 4c). DOPA decarboxylase and DBH-like monooxygenase (which are involved in dopamine, noradrenaline and adrenaline synthesis in adrenergic cells), and putative vesicular amine transporters (which are used for neurotransmitter uptake) are present. The Trichoplax genome encodes members of the synaptic core complex (SNAP-25, synaptobrevin and syntaxin). Whereas synaptobrevin and syntaxin are found in diverse eukaryotic groups and are generally involved in vesicular trafficking, Trichoplax SNAP-25 has a distinct domain found only in animal versions of this protein and not in other eukaryotic members of this family (for example, Sec9 in yeast)41. The genome also contains homologues of the animal neurosecretory vesicle membrane-bound proteins synaptophysin and synaptotagmin, which aid in calcium-dependent vesicle docking and fusion by interacting with SNAP-25.
Putative neurotransmitter and neuropeptide receptors are also present, including abundant seven transmembrane G-protein-coupled receptors (GPCRs) that could be candidate sensory transducers. Four putative opsin genes, which possess a crucial lysine residue in the seventh transmembrane domain and thus are thought to function in light reception, are present. Eighty-five members of the class 3 GPCR family (unrelated to other GPCR families by sequence), including putative metabotropic glutamate receptors, are also found. Transmembrane proteins important in nerve conduction (multiple candidate ionotropic glutamate receptors) and in neurotransmitter release and uptake (for example, sodium neurotransmitter symporter) are encoded by the genome (Fig. 4c). Synapse formation proteins (such as neurexin and neuroligin) and structural elements of the post-synaptic scaffold known to be present in sponges42 (such as discs large) are also found in Trichoplax, including the receptors and channels missing from the sponge genome. Genes associated with neural migration and axon guidance in bilaterians (slit, netrin, NCAM, semaphorin (also known as CD100) and spondin) are also present. Although all of these nominally neural elements may be functioning in non-neural roles, the Trichoplax genome does encode the basic machinery required for the synthesis, release and uptake of neurotransmitters, for synapse formation, and for the conduction of electrical impulses and photoreception.
Extracellular matrix and cell adhesion
Trichoplax has regular cell–cell junctions between epithelial cells but is reported to lack an underlying basal lamina, or indeed, any described extracellular matrix (ECM)2. Its genome, however, contains a diverse set of genes that code for putative ECM proteins (Fig. 4d). These include collagen IV, laminin-α, -β and -γ, and nidogen; however, fibronectin, fibrin, elastin and vitronectin are all apparently absent. Heparan sulphate proteoglycans (including two glypicans) and a matrilin-2-like gene are also found. Because many of these genes were also represented in the ESTs from cultured animals, it is possible that an ECM is present in a way that evades traditional histological stains.
The Trichoplax genome also encodes cell-surface adhesion proteins (α- and β-integrins, cadherins, selectins and immunoglobulin superfamily members) that interact with each other and the ECM in bilaterians, and encodes cytoskeletal linker proteins (paxillin, vinculin, talin and α- and β-catenin) that help organize the actin cytoskeleton and/or transduce signals in other eumetazoans (Fig. 4d). Similarly, protein components (focal adhesion kinase (FAK), paxillin and talin) that would permit dual functions of β-catenin and integrin receptors in adhesion and signal transduction (through Wnt and FAK signalling, respectively) are encoded by the genome. Enzyme families known to modify ECM components and/or signalling molecules in the matrix, such as lysyl oxidases, the ADAM metalloproteases (including the TACE family) and the TIMP metalloprotease inhibitor are also present.
Sex and germ cells
Given the ancient eukaryotic origins of meiosis43, the production of putative oocytes by Trichoplax2, and inference of recombination in wild populations14, it is perhaps not surprising that meiosis-associated genes are found in the genome (Supplementary Table 9.1). Trichoplax has an orthologue of the zinc-finger protein nanos and a member of the vasa/PL10 family of DEAD-box helicases, as well as homologues of mago nashi, PAR-1, pumilio and tudor, which are all implicated in primary germ cell development in eumetazoans. Although these results indicate that Trichoplax has the same genetic tools that cnidarians and bilaterians use to segregate the germ line, further studies to document the expression and functions of these genes are needed to verify germ line formation in placozoans.
Conclusions
Our whole-genome analyses are consistent with placozoans being the earliest diverging eumetazoan phylum, that is, the sister group to the cnidarian–bilaterian clade. Although we cannot formally exclude a more basal position, our analysis rejects the derivation of placozoans from within cnidarians or bilaterians. Further studies (including extra sequences, ideally from whole genomes) will be needed to test this phylogenetic hypothesis.
Although Trichoplax has a compact genome relative to vertebrates and many other animals, we find that it has not experienced the same degree of intron loss and genomic rearrangement as other small (∼100 Mb) metazoan genomes have (for example, the sequences of flies and soil nematodes). This suggests that many structural aspects (introns, local gene order and larger-scale linkages) of the small Trichoplax genome could be primitive eumetazoan characteristics.
Trichoplax’s apparent genomic primitiveness, however, is separate from the question of whether placozoan morphology or life history is a relict of the eumetazoan ancestor. For example, the flat form and gutless feeding could be a ‘primitive’ ancestral feature, with the cnidarian–bilaterian gut arising secondarily by the invention of a developmental process for producing an internal body cavity (as in Bütschli’s ‘plakula’ theory44,45), or it could be a ‘derived’, uniquely placozoan feature that resulted from the loss of an ancestral eumetazoan gut. Unfortunately, the genome sequence alone cannot answer these questions, but it does provide a platform for further studies.
Although the Trichoplax body plan is simple, its genome encodes a rich array of transcription factors and signalling pathways that are typically associated with eumetazoan developmental patterning and cell-type specification. A question remains: what role do these genes have in placozoans? Cellular morphology may be deceptive, and complex gene expression patterns may define functionally distinct but morphologically cryptic cellular subtypes28,31,33. This would be consistent with models in which transcription factors associated with gene expression patterns for specific differentiated cell functions in the eumetazoan ancestor were co-opted in cnidarians and bilaterians for patterning roles46. We speculate that signalling and transcription factor genes may be involved in complex regulatory events required for the known processes of growth, fission and/or swarming, or the as yet undescribed processes of sexual reproduction and embryonic development (Supplementary Fig. 9.1).
It has been suggested that Trichoplax is a ‘living fossil’ relict of an early stage of animal evolution9,44. At least from a genomic perspective, Trichoplax retains many ancestral features of its last common ancestor with cnidarians and bilaterians, which lived in the Precambrian. The extent to which the physiology, behaviour and life history of placozoans retains primitive features remains unclear. With the genome in hand, renewed interest in this ‘simple’ animal with a complex genome will add to our appreciation of animal diversity and perhaps yield fundamental insights into early animal evolution.
Methods Summary
Detailed methods are described in the Supplementary Information. The genome assembly, gene model sequences, predicted proteins and EST clusters and sequences can be downloaded from the JGI website http://www.jgi.doe.gov/trichoplax. Browser display of the genome sequence, including gene predictions and EST and homologous protein alignments, are also available at this site. The sequence data have been deposited in DDBJ/EMBL/GenBank as accession number ABGP00000000.
Online Methods
A detailed description of methods used in this study can be found in the Supplementary Information.
Genome sequencing
Genomic DNA was sheared and cloned into plasmid and fosmid vectors for whole-genome shotgun sequencing as described47. The data were assembled using release 2.10.6 of Jazz, a WGS assembler47. The Trichoplax 8× assembly and the preliminary data analysis can be downloaded from http://www.jgi.doe.gov/trichoplax and has been deposited at DDBJ/EMBL/GenBank under accession number ABGP00000000.
Gene prediction and annotation
The JGI annotation pipeline took scaffolds, repeats and ESTs as inputs and produced gene models and other features that are stored in a relational database. The data can be publicly accessed through the JGI genome portal at http://www.jgi.doe.gov/trichoplax. Protein-coding gene predictions are deposited in DDBJ/EMBL/GenBank as accession ABGP00000000.
Phylogenetic methods
One hundred and four single-copy orthologous genes from nine genomes were aligned using default parameters using both CLUSTALW48 and MUSCLE49, and poorly aligned regions were excluded using Gblocks, yielding 6,783 aligned amino acid positions. Phylogenetic analyses were conducted using Bayesian inference, maximum likelihood with bootstrap, and maximum parsimony with bootstrap using MrBayes50, PHYML51, and PAUP52 respectively. Ribosomal sequences (18S, 5.8S and 28S) were added to the nuclear data set and analysed independently using maximum parsimony with bootstrap. Alternative likelihood topologies were tested using TREEPUZZLE53 and CONSEL54.
Identification of Trichoplax orthologues of specific bilaterian genes
A list of Trichoplax gene models annotated with PANTHER hidden Markov models55 or PFAM domains56 was analysed for genes involved in various biological processes in bilaterians. In many cases, Trichoplax orthologues of bilaterian genes were identified by BLAST against the Trichoplax assembly. The resulting genes were analysed by BLAST against the database of non-redundant proteins, PFAM domain composition and phylogenetic trees to determine orthology to the vertebrate query.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
This paper is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence, and is freely available to all readers at http://www.nature.com/nature. The sequences generated in this study are available from GenBank under the accession number ABGP00000000.
References
Schulze, F. E. Trichoplax adhaerens, nov. gen., nov. spec. Zool. Anz. 6, 92–97 (1883)
Grell, K. G. & Ruthmann, A. in Placozoa, Porifera, Cnidaria and Ctenophora (eds Harrison, F. W. & Westfall, J. A.) 13–27 (Wiley-Liss, 1991)
Pearse, V. B. Growth and behavior of Trichoplax adhaerens: first record for the phylum Placozoa in Hawaii. Pacif. Sci. 43, 117–121 (1989)
Maruyama, Y. K. Occurrence in the field of a long-term, year-round, stable population of placozoans. Biol. Bull. 206, 55–60 (2004)
Voigt, O. et al. Placozoa—no longer a phylum of one. Curr. Biol. 14, R944–R945 (2004)
Pearse, V. B. & Voigt, O. Field biology of placozoans (Trichoplax): distribution, diversity, biotic interactions. Integr. Comp. Biol. 47, 677–692 (2007)
Signorovitch, A. Y., Dellaporta, S. L. & Buss, L. W. Caribbean placozoan phylogeography. Biol. Bull. 211, 149–156 (2006)
Grell, K. G. Trichoplax adhaerens, F.E. Schulze und die Entstehung der Metazoen. Naturwiss. Rundsch. 24, 160–161 (1971)
Schierwater, B. My favorite animal, Trichoplax adhaerens . BioEssays 27, 1294–1302 (2005)
Ruthmann, A., Behrendt, G. & Wahl, R. The ventral epithelium of Trichoplax adhaerens (Placozoa): Cytoskeletal structures, cell contacts and endocytosis. Zoomorphology 106, 115–112 (1986)
Wenderoth, H. Transepithelial cytophagy by Trichoplax adhaerens F. E. Schulze (Placozoa) feeding on yeast. Z. Naturforsch. 41C, 343–347 (1986)
Grell, K. G. Eibildung und Furchung von Trichoplax adhaerens F. E. Schulze (Placozoa). Z. Morph. Tiere 73, 297–314 (1972)
Grell, K. G. & Benwitz, G. Ergänzende untersuchungen zur ultrastruktur von Trichoplax adhaerens F.E. Schulze (Placozoa). Zoomorphology 98, 47–67 (1981)
Signorovitch, A. Y., Dellaporta, S. L. & Buss, L. W. Molecular signatures for sex in the Placozoa. Proc. Natl Acad. Sci. USA 102, 15518–15522 (2005)
Bridge, D., Cunningham, C. W., DeSalle, R. & Buss, L. W. Class-level relationships in the phylum Cnidaria: molecular and morphological evidence. Mol. Biol. Evol. 12, 679–689 (1995)
Collins, A. G. Evaluating multiple alternative hypotheses for the origin of Bilateria: an analysis of 18S rRNA molecular evidence. Proc. Natl Acad. Sci. USA 95, 15458–15463 (1998)
Ender, A. & Schierwater, B. Placozoa are not derived cnidarians: evidence from molecular morphology. Mol. Biol. Evol. 20, 130–134 (2003)
da Silva, F. B., Muschner, V. C. & Bonatto, S. L. Phylogenetic position of Placozoa based on large subunit (LSU) and small subunit (SSU) rRNA genes. Genet. Mol. Biol. 30, 127–132 (2007)
Dellaporta, S. L. et al. Mitochondrial genome of Trichoplax adhaerens supports placozoa as the basal lower metazoan phylum. Proc. Natl Acad. Sci. USA 103, 8751–8756 (2006)
Signorovitch, A. Y., Buss, L. W. & Dellaporta, S. L. Comparative genomics of large mitochondria in placozoans. PLoS Genet. 3, e13 (2007)
Ruiz-Trillo, I., Roger, A. J., Burger, G., Gray, M. W. & Lang, B. F. A phylogenomic investigation into the origin of Metazoa. Mol. Biol. Evol. 25, 664–672 (2008)
Weber, J. L. & Myers, E. W. Human whole-genome shotgun sequencing. Genome Res. 7, 401–409 (1997)
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007)
Haen, K. M., Lang, B. F., Pomponi, S. A. & Lavrov, D. V. Glass sponges and bilaterian animals share derived mitochondrial genomic features: a common ancestry or parallel evolution? Mol. Biol. Evol. 24, 1518–1527 (2007)
Lavrov, D. V., Forget, L., Kelly, M. & Lang, B. F. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol. Biol. Evol. 22, 1231–1239 (2005)
Renwick, J. H. The mapping of human chromosomes. Annu. Rev. Genet. 5, 81–120 (1971)
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotope. Nature 453, 1064–1071 (2008)
Jakob, W. et al. The Trox-2 Hox/ParaHox gene of Trichoplax (Placozoa) marks an epithelial boundary. Dev. Genes Evol. 214, 170–175 (2004)
Hadrys, T., DeSalle, R., Sagasser, S., Fischer, N. & Schierwater, B. The Trichoplax PaxB gene: a putative Proto-PaxA/B/C gene predating the origin of nerve and sensory cells. Mol. Biol. Evol. 22, 1569–1578 (2005)
Monteiro, A. S., Schierwater, B., Dellaporta, S. L. & Holland, P. W. A low diversity of ANTP class homeobox genes in Placozoa. Evol. Dev. 8, 174–182 (2006)
Martinelli, C. & Spring, J. Expression pattern of the homeobox gene Not in the basal metazoan Trichoplax adhaerens . Gene Expr. Patterns 4, 443–447 (2004)
Schierwater, B. et al. The ancestral Antp gene repertoire: Insights from the placozoan genome. PLoS ONE (in the press)
Martinelli, C. & Spring, J. Distinct expression patterns of the two T-box homologues Brachyury and Tbx2/3 in the placozoan Trichoplax adhaerens . Dev. Genes Evol. 213, 492–499 (2003)
Lee, P. N., Kumburegama, S., Marlow, H. Q., Martindale, M. Q. & Wikramanayake, A. H. Asymmetric developmental potential along the animal–vegetal axis in the anthozoan cnidarian, Nematostella vectensis, is mediated by Dishevelled. Dev. Biol. 310, 169–186 (2007)
Adamska, M. et al. Wnt and TGF-β expression in the sponge Amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS ONE 2, e1031 (2007)
Matus, D. Q., Thomsen, G. H. & Martindale, M. Q. Dorso/ventral genes are asymmetrically expressed and involved in germ-layer demarcation during cnidarian gastrulation. Curr. Biol. 16, 499–505 (2006)
Adamska, M. et al. The evolutionary origin of Hedgehog proteins. Curr. Biol. 17, R836–R837 (2007)
Nichols, S. A., Dirks, W., Pearse, J. S. & King, N. Early evolution of animal cell signaling and adhesion genes. Proc. Natl Acad. Sci. USA 103, 12451–12456 (2006)
Muller, W. E. & Schacke, H. Characterization of the receptor protein-tyrosine kinase gene from the marine sponge Geodia cydonium . Prog. Mol. Subcell. Biol. 17, 183–208 (1996)
Schuchert, P. Trichoplax adhaerens (phylum Placozoa) has cells that react with antibodies against the neuropeptide RFamide. Acta Zool. 74, 115–117 (1993)
Risinger, C. et al. Evolutionary conservation of synaptosome-associated protein 25 kDa (SNAP-25) shown by Drosophila and Torpedo cDNA clones. J. Biol. Chem. 268, 24408–24414 (1993)
Sakarya, O. et al. A post-synaptic scaffold at the origin of the animal kingdom. PLoS ONE 2, e506 (2007)
Ramesh, M. A., Malik, S. B. & Logsdon, J. M. A phylogenomic inventory of meiotic genes; evidence for sex in Giardia and an early eukaryotic origin of meiosis. Curr. Biol. 15, 185–191 (2005)
Bütschli, O. Bemerkungen zur Gastraeatheorie. Morph. Jahrb. 9, 415–427 (1884)
Syed, T. & Schierwater, B. The evolution of the Placozoa: a new morphological model. Senckenbergiana Lethaea 82, 259–270 (2002)
Erwin, D. H. & Davidson, E. H. The last common bilaterian ancestor. Development 129, 3021–3032 (2002)
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes . Science 297, 1301–1310 (2002)
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994)
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004)
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003)
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003)
Swofford, D. L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), version 4.0. (Sinauer Associates, 1999)
Schmidt, H. A., Strimmer, K., Vingron, M. & von Haeseler, A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502–504 (2002)
Shimodaira, H. & Hasegawa, M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (2001)
Thomas, P. D. et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003)
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004)
Acknowledgements
This work was performed under the auspices of the USA Department of Energy’s Office of Science, Biological and Environmental Research Program and by the University of California, Lawrence Livermore National Laboratory, Lawrence Berkeley National Laboratory and Los Alamos National Laboratory. The Center for Integrative Genomics is supported by a grant from the Gordon and Betty Moore Foundation. D.S.R. acknowledges support from R. A. Melmon. L.W.B., S.L.D. and M.A.M. were supported by the National Science Foundation. B.S. acknowledges support from the German Science Foundation. S.L.D. and B.S. also acknowledge support from Human Frontiers Science Program.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
The file contains extensive Supplementary Information which includes Supplementary Data, Supplementary Notes, Supplementary Figures (S2.1-S9.18) and Supplementary Tables (S2.1-S9.3). (PDF 4587 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Srivastava, M., Begovic, E., Chapman, J. et al. The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 (2008). https://doi.org/10.1038/nature07191
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature07191
This article is cited by
-
The placozoan Trichoplax
Nature Methods (2024)
-
On being a Hydra with, and without, a nervous system: what do neurons add?
Animal Cognition (2023)
-
Emergence of distinct syntenic density regimes is associated with early metazoan genomic transitions
BMC Genomics (2022)
-
A developmental role for the chromatin-regulating CoREST complex in the cnidarian Nematostella vectensis
BMC Biology (2022)
-
Mass spectrometry of short peptides reveals common features of metazoan peptidergic neurons
Nature Ecology & Evolution (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
