
Abstract
Choanoflagellates are the closest known relatives of metazoans. To discover potential molecular mechanisms underlying the evolution of metazoan multicellularity, we sequenced and analysed the genome of the unicellular choanoflagellate Monosiga brevicollis. The genome contains approximately 9,200 intron-rich genes, including a number that encode cell adhesion and signalling protein domains that are otherwise restricted to metazoans. Here we show that the physical linkages among protein domains often differ between M. brevicollis and metazoans, suggesting that abundant domain shuffling followed the separation of the choanoflagellate and metazoan lineages. The completion of the M. brevicollis genome allows us to reconstruct with increasing resolution the genomic changes that accompanied the origin of metazoans.
Similar content being viewed by others

Main
Choanoflagellates have long fascinated evolutionary biologists for their marked similarity to the ‘feeding cells’ (choanocytes) of sponges and the possibility that they might represent the closest living relatives of metazoans1,2. Over the past decade or so, evidence supporting this relationship has accumulated from phylogenetic analyses of nuclear and mitochondrial genes3,4,5,6, comparative genomics between the mitochondrial genomes of choanoflagellates, sponges and other metazoans7,8, and the finding that choanoflagellates express homologues of metazoan signalling and adhesion genes9,10,11,12. Furthermore, species-rich phylogenetic analyses demonstrate that choanoflagellates are not derived from metazoans, but instead represent a distinct lineage that evolved before the origin and diversification of metazoans (Fig. 1a, Supplementary Fig. 1 and Supplementary Note 3.1)8,13. By virtue of their position on the tree of life, studies of choanoflagellates provide an unparallelled window into the nature of the unicellular and colonial progenitors of metazoans14.

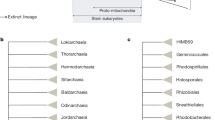
a, The close phylogenetic affinity between choanoflagellates and metazoans highlights the value of the M. brevicollis genome for investigations into metazoan origins, the biology of the last common ancestor of metazoans (filled circle) and the biology of the last common ancestor of choanoflagellates and metazoans (open circle). Genomes from species shown with their abbreviation were used for protein domain comparisons in this study: human (Homo sapiens, Hsap), ascidian (Ciona intestinalis, Cint), Drosophila (Drosophila melanogaster, Dmel), cnidarian (N. vectensis, Nvec), M. brevicollis (Mbre), zygomycete (Rhizopus oryzae, Rory), basidiomycete (Coprinus cinereus, Ccin), ascomycete (Neurospora crassa, Ncra), hemiascomycete (Saccharomyces cerevisiae, Scer), slime mould (Dictyostelium discoideum, Ddis) and Arabidopsis (Arabidopsis thaliana, Atha). b–d, Choanoflagellate cells bear a single apical flagellum (arrow, b) and an apical collar of actin-filled microvilli (bracket, c). d, An overlay of β-tubulin (green), polymerized actin (red) and DNA localization (blue) reveals the position of the flagellum within the collar of microvilli. Scale bar, 2 μm.
Choanoflagellates are abundant and globally distributed microbial eukaryotes found in marine and freshwater environments15,16. Like sponge choanocytes, each cell bears an apical flagellum surrounded by a distinctive collar of actin-filled microvilli, with which choanoflagellates trap bacteria and detritus (Fig. 1b). Using this highly effective means of prey capture, choanoflagellates link bacteria to higher trophic levels and thus have critical roles in oceanic carbon cycling and in the microbial food web17,18.
More than 125 choanoflagellate species have been identified, and all species have a unicellular life-history stage. Some can also form simple colonies of equipotent cells, although these differ substantially from the obligate associations of differentiated cells in metazoans19. Studies of basal metazoans indicate that the ancestral metazoan was multicellular and had differentiated cell types, an epithelium, a body plan and regulated development including gastrulation. In contrast, the last common ancestor of choanoflagellates and metazoans was unicellular or possibly capable of forming simple colonies, underscoring the abundant biological innovation that accompanied metazoan origins.
Despite their evolutionary and ecological importance, little is known about the genetics and cell biology of choanoflagellates. To gain insight into the biology of choanoflagellates and to reconstruct the genomic changes attendant on the early evolution of metazoans, we sequenced the genome of the choanoflagellate M. brevicollis and compared it with genomes from metazoans and other eukaryotes.
Gene structure and intron evolution
The ∼41.6 megabase (Mb) M. brevicollis genome contains approximately 9,200 genes (Supplementary Notes 1 and 2) and is comparable in size to the genomes of filamentous fungi (∼30–40 Mb) and other free-living unicellular eukaryotes (for example, small diatoms at ∼20–35 Mb20 and ichthyosporeans at ∼20–25 Mb21). Metazoan genomes are typically larger, with few exceptions22.
M. brevicollis genes have several distinguishing structural features (Table 1). Whereas the M. brevicollis genome is compact, its genes are almost as intron-rich as human genes (6.6 introns per M. brevicollis gene versus 7.7 introns per human gene). M. brevicollis introns are short (averaging 174 bp) relative to metazoan introns, and with few exceptions do not include the extremely long introns found in some metazoan genes (Supplementary Fig. 2 and Supplementary Note 3.3).
Comparisons of intron positions in a set of conserved genes from M. brevicollis, diverse metazoans and representative intron-rich fungi, plants and a ciliate reveal that the last common ancestor of choanoflagellates and metazoans had genes at least as intron-rich as those of modern choanoflagellates (Fig. 2, Supplementary Figs 3 and 4, and Supplementary Note 3.3). Notably, these analyses reveal that the eumetazoan ancestor contained a substantially higher density of introns than the last common ancestor of choanoflagellates and metazoans. This is consistent with a proliferation of introns during the early evolution of the Metazoa23.

Ancestral intron content, intron gains and intron losses were inferred by the Csuros maximum likelihood method45 from a sample of 1,054 intron positions in 473 highly conserved genes in representative metazoans (humans, D. melanogaster and N. vectensis), M. brevicollis, intron-rich fungi (Cryptococcus neoformans A and Phanerochaete chrysosporium), plants and green algae (A. thaliana and Chlamydomonas reinhardtii), and a ciliate (Tetrahymena thermophila). Branches with more gain than loss are blue, those with more loss than gain are red, and those with comparable amounts of each are black. The inferred or observed number of introns present in ancestral and extant species is indicated by proportionally sized circles. As in Fig. 1, the last common ancestor of metazoans and the last common ancestor of choanoflagellates and metazoans are represented by a filled circle and an open circle, respectively. Other ancestral nodes are indicated by grey circles.
Premetazoan history of protein domains required for multicellularity
The M. brevicollis genome provides unprecedented insight into the early evolution of metazoan genes. Annotation of the M. brevicollis genome using Pfam and SMART (two protein domain prediction databases) reveals 78 protein domains that are exclusive to choanoflagellates and metazoans, only two of which have been reported previously in choanoflagellates (Supplementary Table 4)10. Because genomic features shared by M. brevicollis and metazoans were probably present in their last common ancestor, this study extends the evolutionary history of a cohort of important protein domains to the premetazoan era. Many of these domains are central to cell signalling and adhesion processes in metazoans, suggesting a role in the origin of multicellularity. In contrast, metazoan genomic features that are missing from the M. brevicollis genome may have evolved within the metazoan lineage, or may have existed in the last common ancestor with choanoflagellates and were subsequently lost on the stem leading to M. brevicollis. Presumably, there are many genomic features that evolved in the metazoan lineage, and the M. brevicollis genome provides our first glimpse at the complement of genes and protein domains that predate metazoan origins.
To investigate further the extent to which molecular components required for metazoan multicellularity evolved before the origin of metazoans, we performed targeted searches in the M. brevicollis genome and representative metazoan, fungal and plant genomes for homologues of critical metazoan cell adhesion, cell signalling and transcription factor protein families.
An abundance of cell adhesion domains
A critical step in the transition to multicellularity was the evolution of mechanisms for stable cell adhesion. M. brevicollis encodes a diverse array of cell adhesion and extracellular matrix (ECM) protein domains previously thought to be restricted to metazoans (Fig. 3). At least 23 M. brevicollis genes encode one or more cadherin domains, homologues of which are required for cell sorting and adhesion during metazoan embryogenesis24, and 12 genes encode C-type lectins, 2 of which are transmembrane proteins. Whereas soluble C-type lectins have functions ranging from pathogen recognition to ECM organization, transmembrane C-type lectins mediate specific adhesive activities such as contact between leukocytes and vascular endothelial cells, cell recognition, and molecular uptake by endocytosis25,26,27.

M. brevicollis possesses diverse adhesion and ECM domains previously thought to be unique to metazoans (magenta). In contrast, many metazoan sequence-specific transcription factors are absent from the M. brevicollis gene catalogue. For adhesion and ECM domains, a filled box indicates a domain identified by both SMART and Pfam37,48, a half-filled box indicates a domain identified by either SMART or Pfam, and an open box indicates a domain that is not encoded by the current set of gene models. The presence (filled box) or absence (empty box) of transcription factor families was determined by reciprocal BLAST and SMART/Pfam domain annotations (Supplementary Note 3.5). Species names follow the convention from Fig. 1. EC, extracellular domain; cyto, cytoplasmic domain; asterisk, collagen triple-helix-domains occur in the extended tandem arrays diagnostic of collagen proteins found only in metazoans and choanoflagellates.
The genome of M. brevicollis also contains integrin-α and immunoglobulin domains—cell adhesion domains formerly thought to be restricted to metazoans. In metazoans, integrin-α- and integrin-β-domain-containing proteins heterodimerize before binding to ECM proteins such as collagen28. We find that M. brevicollis has at least 17 integrin-α-domain-containing proteins, but no integrin-β domains. Metazoan immunoglobulin-domain-containing proteins have both adhesive and immune functions. The M. brevicollis genome encodes a total of five immunoglobulin domains that show affinity for the I-set, V-set or C2-set subfamilies, but not the vertebrate-specific C1-set subfamily. In contrast to M. brevicollis, metazoan genomes possess from ∼150 to ∼1,500 immunoglobulin domains (Supplementary Table 7), suggesting that the radiation of the immunoglobulin superfamily occurred after the divergence of choanoflagellates and metazoans.
The finding in M. brevicollis of cell adhesion domains that were previously known only in metazoans has two important implications. First, the common ancestor of metazoans and choanoflagellates possessed several of the critical structural components used for multicellularity in modern metazoans. Second, given the absence of evidence for stable cell adhesion in M. brevicollis, this also suggests that homologues of metazoan cell adhesion domains may act to mediate interactions between M. brevicollis and its extracellular environment.
Extracellular-matrix-associated protein domains
As the targets of many adhesion receptors, the question of whether metazoan-type ECM proteins and domains evolved before or after the transition to multicellularity is of great interest. In metazoans, collagens are ECM proteins that polymerize to form a major component of the basement membrane of epithelia and have been invoked as a potential ‘key innovation’ during the transition to multicellularity29. We find five collagen-domain-encoding genes in the M. brevicollis genome, two of which encode the diagnostic Gly-X-Y repetitive sequence motif (where X and Y are frequently proline and hydroxyproline, respectively) in an arrangement similar to metazoan collagens30. Other ECM-associated domains previously known only from metazoans that occur in M. brevicollis include laminin domains (an important class that contributes to the basement membrane), the reeler domain (found in the neuronal ECM protein reelin31) and the ependymin domain (an extracellular glycoprotein found in cerebrospinal fluid32; Fig. 3 and Supplementary Table 4).
The discovery of putatively secreted ECM proteins in a free-living choanoflagellate suggests that elements of the metazoan ECM evolved in contact with the external environment before being sequestered within an epithelium. Although some choanoflagellates secrete extracellular structures or adhere to form colonial assemblages19,33,34, M. brevicollis is not known to do so. Instead, these ECM protein homologues in M. brevicollis may mediate an analogous process such as substrate attachment.
Against the backdrop of abundant conservation of cell adhesion and ECM protein domains among the genomes of M. brevicollis and metazoans, it is important to note the differences. Individual cell adhesion and ECM-associated domains in the M. brevicollis genome often occur in unique arrangements, and clear orthologues of specific metazoan adhesion proteins are rarely found. Although the domains associated with metazoan adhesion and ECM proteins were present in the ancestor of choanoflagellates and metazoans, the canonical metazoan adhesion protein architectures35 probably evolved after the divergence of the two lineages.
Domain shuffling in the evolution of intercellular signalling pathways
Our analysis of the M. brevicollis genome reveals little evidence that metazoan-specific signalling pathways were present in the last common ancestor of choanoflagellates and metazoans. Many pathways are missing entirely, and M. brevicollis genes with some similarity to metazoan signalling machinery are largely found to share conserved domains without aligning across the full span of what are often complex multidomain proteins (for example, epidermal growth factor (EGF) repeats are common to Notch, and also to many other proteins; Supplementary Table 8). Specifically, no receptors or ligands were identified from the NHR (nuclear hormone receptor), WNT and TGF-β signalling pathways. The only evidence of the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway is an apparent STAT-like gene that encodes a STAT DNA-binding domain and a partial SH2 domain. Convincing evidence is also lacking for the Toll signalling pathway—a signalling system important both for development and for innate immunity in metazoans.
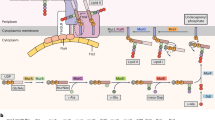
Nonetheless, the genome of M. brevicollis does provide insights into the evolution of Notch and hedgehog (Hh) signalling pathways. Cassettes of protein domains found in metazoan Notch receptors (EGF, NL and ANK (ankyrin repeats)) are encoded on separate M. brevicollis genes in arrangements that differ from metazoan Notch proteins, and definitive domains, such as the NOD domain and the MNNL region, are absent (Fig. 4a).

Analysis of the draft gene set reveals that M. brevicollis possesses proteins containing domains characteristic of metazoan Notch (a, N1–N3) and hedgehog (b, H1 and H2). Some of these protein domains were previously thought to be unique to metazoans. The presence of these domains in separate M. brevicollis proteins implicates domain shuffling in the evolution of Notch and Hedgehog. Hh, hedgehog; N-hh, hedgehog N-terminal signalling domain; Hint, hedgehog/intein domain; TM, transmembrane domain; VWA, von Willebrand A domain. See Supplementary Note 3.6 for protein accession numbers and Supplementary Fig. 6 for identification of all displayed protein domains. Species names follow the convention from Fig. 1.
Homologues of hedgehog, dispatched and patched genes are also present; however, there is no evidence for smoothened nor for its defining frizzled domain. In metazoans, hedgehog consists of an amino-terminal signalling domain and carboxy-terminal hedgehog/intein (Hint) domain responsible for autocatalytically cleaving the protein. In one M. brevicollis hedgehog-like protein, a hedgehog N-terminal signalling domain is found at the N terminus of a large transmembrane protein that, instead of a Hint domain, includes von Willebrand A, cadherin, TNFR (tumor necrosis factor receptor), furin and EGF domains. Similar proteins are found in the sponge Amphimedon queenslandica and in the cnidarian Nematostella vectensis36, revealing that the M. brevicollis genome captures an ancestral arrangement of protein domains rather than representing a lineage-specific domain-shuffling event. Another M. brevicollis hedgehog-like protein contains a Hint domain—a key region involved in the autocatalytic processing of hedgehog (Fig. 4b). The identification of a hedgehog-like gene in a choanoflagellate is not without precedent. A distinct Hint-domain-containing protein, named Hoglet, was identified in the distantly related Monosiga ovata12, supporting the idea that isolated signalling components were present in the last common ancestor of choanoflagellates and metazoans.
Divergent use of phosphotyrosine signalling machinery
Phosphotyrosine (pTyr)-based signalling was considered unique to metazoans until its recent discovery in choanoflagellates9,11. The key domains involved in pTyr signalling are found in abundance in the M. brevicollis genome: tyrosine kinase domains that phosphorylate tyrosine (∼120 occurrences), pTyr-specific phosphatases (PTP) that remove the phosphate modification (∼30) and SH2 domains that bind pTyr-containing peptides (∼80) (Supplementary Fig. 7). In contrast, these domains are rare in non-metazoans; for example, S. cerevisiae has no tyrosine kinase domains, only three PTP domains and a single SH2 domain. These findings support a model in which the full set of pTyr signalling machinery evolved before the separation of the choanoflagellate and metazoan lineages.
Although pTyr signalling machinery is present in metazoans and choanoflagellates, the mode of usage in M. brevicollis may be distinct from that in metazoans. A simple metric for the use of a particular domain is the range of domain types with which it is found in combination37. In the M. brevicollis genome, more than half of the observed pairwise domain combinations involving tyrosine kinase, PTP and SH2 domains are distinct from those seen in any metazoan genome (Fig. 5 and Supplementary Note 3.7). In contrast, for other sets of common signalling domains (those involved in phosphoserine/threonine (pSer/Thr), Ras–GTP and Rho–GTP signalling), most observed combinations are shared between M. brevicollis and metazoans. These observations are consistent with a simple model in which pSer/Thr, Ras–GTP and Rho–GTP signalling were fully elaborated before the branching of the choanoflagellate and metazoan lineages (consistent with the presence of these systems in other eukaryotes, including fungi, Dictyostelium and plants). In contrast, simple pTyr signalling may have emerged in the common ancestor and diverged radically between choanoflagellates and metazoans.

A metric for functional usage of a domain within a genome is the number of other domains with which it co-occurs in a single protein. Numbers of pairwise domain combinations are indicated for classes of signalling domains involved in Ras, Rho, pSer/Thr and pTyr signalling. In cases in which a domain combination occurs multiple times within an individual protein or genome, it is only counted once. All combinations observed in M. brevicollis are indicated either as those that are only observed in the M. brevicollis genome (magenta) or as those that are observed both in M. brevicollis and metazoan genomes (grey). pTyr signalling domains in M. brevicollis are unique in that most of their observed pairwise domain combinations are distinct from those observed in metazoans. GEF, guanine-nucleotide exchange factor; GAP, GTPase-activating protein.
Streamlined transcriptional regulation
The core transcriptional apparatus of M. brevicollis is, in many ways, typical of most eukaryotes examined to date (Supplementary Table 10) including, for example, all 12 RNA polymerase II subunits and most of the transcription elongation factors (TFIIS, NELF, PAF, DSIF and P-TEFb, but not elongin). However, homologues of the largest subunit of TFIIF and several subunits of TFIIH are apparently lacking from the genome and the expressed-sequence-tag collection (Supplementary Fig. 8), reminiscent of the absence of several basal factors from the Giardia lamblia genome, suggesting alternative strategies for interacting with core promoter elements38. Similarly, only a limited number of general co-activators are identifiable in M. brevicollis, including the components of several chromatin-remodelling complexes (Supplementary Fig. 9 and Supplementary Note 3.8).
Perhaps not surprisingly, M. brevicollis possesses members from most of the ubiquitous families of eukaryotic transcription factors (Supplementary Fig. 10). Most of the predicted transcription factors are zinc-coordinating; approximately 44% are C2H2-type zinc fingers. Eight proteins (5% of a total of 155 predicted transcription factors) are forkhead transcription factors, otherwise known only from metazoans and fungi.
The homeodomain transcription factors are an ancient protein family found in all known eukaryotes. At least two major superclasses of homeodomain proteins evolved before the origin of metazoans: those containing homeodomains of 60 amino acids (the ‘typical’, or non-TALE superclass), and those containing homeodomains of more than 63 amino acids (the TALE superclass)39. The M. brevicollis genome encodes only two homeodomain proteins, both of which group with the MEIS sub-class of TALE homeodomains (Supplementary Fig. 12). Apparently, genes encoding non-TALE homeodomain proteins have been lost in the lineage leading to M. brevicollis. Bona fide HOX class homeobox genes—a subclass of the non-TALE superclass—are absent from both M. brevicollis and the Amphimedon queenslandica (demosponge) genome sequence reads, indicating that this characteristic metazoan gene family probably emerged along the stem leading to eumetazoans40.
M. brevicollis contains a subset of the transcription factor families previously thought to be specific to metazoans. Members of the metazoan p53, Myc and Sox/TCF families were identified, whereas many transcription factor families associated with metazoan patterning and development (ETS, HOX, NHR, POU and T-box) seem to be absent (Fig. 3).
Discussion
Choanoflagellates, sponges and other metazoans last shared a unicellular common ancestor in the late Precambrian period, more than 600 million years ago41,42. Although the origin of metazoans was a pivotal event in life’s history, little is known about the genetic underpinnings of the requisite transition to multicellularity. Comparisons of modern genomes provide our most direct insights into the ancient genomic conditions from which metazoans emerged. By comparing choanoflagellate and metazoan genomes, we infer that their common ancestor had intron-rich genes, some of which encoded protein domains characteristically associated with cell adhesion and the ECM in animals.
In addition to containing protein domains associated with metazoan cell adhesion, M. brevicollis possesses a surprising abundance of tyrosine kinases and their downstream signalling targets. In contrast, components of most other intercellular signalling pathways, as well as many of the diverse transcription factors that comprise the developmental toolkit of modern animals, are absent. These presumably reached their modern form on the metazoan stem, although it is formally possible that they were in place much earlier and degenerated in the M. brevicollis lineage. Likewise, it is possible that the last common ancestor of choanoflagellates and metazoans had an early form of multicellularity that became more robust in metazoans and was lost in the choanoflagellate lineage. In any event, the evolutionary distance between choanoflagellates and metazoans is substantial, and evidently few, if any, intermediate lineages survived. There are, for example, no other known microbial eukaryotes that possess any of the eight developmental signalling pathways characteristic of metazoans.
The mechanism of invention of new genes on the metazoan stem, and their integration to create the emergent network of cell signalling and transcriptional regulation fundamental to metazoan biology, remains mysterious. Domain shuffling, which has frequently been proposed as an important mechanism for the evolution of metazoan multidomain proteins43,44, is implicated by the presence of essential metazoan signalling domains in M. brevicollis that appear in unique combinations relative to animals. For pTyr-based signalling in particular, the marked divergence of domain combinations suggests that this mode of cellular interaction existed in a nascent form in the common choanoflagellate–metazoan ancestor, and was subsequently specialized and elaborated on in each lineage.
Given the limited transcription factor diversity in M. brevicollis, it is notable that the genome encodes representatives of the otherwise metazoan-specific p53, Myc and Sox/TCF transcription factor families. These transcription factors may have had early and critical roles in the evolution of metazoan ancestors by regulating the differential expression of genes to allow multiple cell types to exist in a single organism, and their study in choanoflagellates is a promising future direction.
The M. brevicollis sequence opens the door to genome-enabled studies of choanoflagellates, a diverse group of microbial eukaryotes that are important in their own right as bacterial predators in both marine and freshwater ecosystems. Although M. brevicollis is strictly unicellular, other choanoflagellates facultatively form colonies, and the modulation of these associations by cell signalling, adhesion, transcriptional regulation and environmental influences is poorly understood. An integrative approach that unites studies of choanoflagellate genomes, cell biology and ecology with the biogeochemistry of the Precambrian promises to reveal the intrinsic and extrinsic factors underlying metazoan origins.
Methods Summary
All analyses described were performed on Version 1.0 of the genome sequence. Details can be found in the Supplementary Information.
Separation of choanoflagellate and bacterial DNA
Using physical separation techniques combined with antibiotic treatments, a culture line with only a single bacterial food source, Flavobacterium sp., was developed. The GC content of Flavobacterium DNA (33%) is sufficiently different from that of M. brevicollis (55%) to allow separation over a CsCl gradient. M. brevicollis genomic DNA was used to construct replicate libraries containing inserts of 2–3 kilobases (kb), 6–8 kb and 35–40 kb.
Genome sequencing, assembly and validation
The draft sequence of the M. brevicollis genome was generated from ∼8.5-fold redundant paired-end whole-genome shotgun sequence coverage (Supplementary Information). Sequence data derived from six whole-genome shotgun libraries were assembled using release 2.9.2 of the whole-genome shotgun assembler Jazz. Completeness of the draft genome was assessed by capturing ∼98.5% of sequenced expressed sequence tags.
Gene prediction and annotation
Gene models (9,196) were predicted and annotated using the Joint Genome Institute (JGI) Annotation Pipeline (Supplementary Information).
Intron analysis
Homologues of 473 highly conserved genes from M. brevicollis and representative eukaryotes were aligned to reveal the position and phylogenetic distribution of 1,989 highly reliable intron splice sites at 1,054 conserved positions. The evolutionary history of introns in orthologous genes was inferred using Dollo parsimony, Roy-Gilbert maximum likelihood and Csuros maximum likelihood45,46,47.
Analysis of signalling, adhesion and transcription factor protein domains
Gene models containing metazoan signalling, adhesion and transcription factor domains were identified by text and protein domain ID searches of the JGI M. brevicollis genome portal, local BLAST searches within the M.brevicollis genome scaffolds, the online Pfam and SMART tools, and reciprocal BLAST searches in the NCBI non-redundant protein database (Supplementary Information).
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The sequenced strain of M. brevicollis has been deposited at http://ATCC.org under accession number PRA-258. The genome assembly and annotation data are deposited at DBJ, EMBL and GenBank under the project accession ABFJ00000000.
References
James-Clark, H. On the spongiae ciliatae as infusoria flagellata; or observations on the structure, animality, and relationship of Leucosolenia botryoides . Ann. Mag. Nat. His. 1, 133–142, 188–215. 250–264 (1868)
Saville Kent, W. A Manual of the Infusoria (Bogue, London, 1880–1882)
Steenkamp, E. T., Wright, J. & Baldauf, S. L. The protistan origins of animals and fungi. Mol. Biol. Evol. 23, 93–106 (2006)
Medina, M. et al. Phylogeny of Opisthokonta and the evolution of multicellularity and complexity in Fungi and Metazoa. Int. J. Astrobiology 2, 203–211 (2003)
Philippe, H. et al. Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol. Biol. Evol. 21, 1740–1752 (2004)
Lang, B. F., O’Kelly, C., Nerad, T., Gray, M. W. & Burger, G. The closest unicellular relatives of animals. Curr. Biol. 12, 1773–1778 (2002)
Burger, G., Forget, L., Zhu, Y., Gray, M. W. & Lang, B. F. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc. Natl Acad. Sci. USA 100, 892–897 (2003)
Lavrov, D. V., Forget, L., Kelly, M. & Lang, B. F. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol. Biol. Evol. 22, 1231–1239 (2005)
King, N. & Carroll, S. B. A receptor tyrosine kinase from choanoflagellates: molecular insights into early animal evolution. Proc. Natl Acad. Sci. USA 98, 15032–15037 (2001)
King, N., Hittinger, C. T. & Carroll, S. B. Evolution of key cell signaling and adhesion protein families predates animal origins. Science 301, 361–363 (2003)
Segawa, Y. et al. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc. Natl Acad. Sci. USA 103, 12021–12026 (2006)
Snell, E. A. et al. An unusual choanoflagellate protein released by Hedgehog autocatalytic processing. Proc. Biol. Sci. 273, 401–407 (2006)
Rokas, A., Kruger, D. & Carroll, S. B. Animal evolution and the molecular signature of radiations compressed in time. Science 310, 1933–1938 (2005)
King, N. The unicellular ancestry of animal development. Dev. Cell 7, 313–325 (2004)
Buck, K. R. & Garrison, D. L. Distribution and abundance of choanoflagellates (Acanthoecidae) across the ice-edge zone in the Weddell Sea, Antarctica. Mar. Biol. 98, 263–269 (1988)
Thomsen, H. A. & Larsen, J. Loricate choanoflagellates of the Southern Ocean with new observations on cell-division in Bicosta spinifera (Throndsen, 1970) from Antarctica and Saroeca attenuata Thomsen, 1979, from the Baltic Sea. Polar Biol. 12, 53–63 (1992)
Arndt, H. et al. in The Flagellates (ed. Leadbeater, B. S. C. & Green, J. C.) 240–268 (Taylor & Francis, London, 2000)
Boenigk, J. & Arndt, H. Bacterivory by heterotrophic flagellates: community structure and feeding strategies. Antonie Van Leeuwenhoek 81, 465–480 (2002)
Leadbeater, B. S. C. Life-history and ultrastructure of a new marine species of Proterospongia (Choanoflagellida). J. Mar. Biol. Assoc.UK 63, 135–160 (1983)
Armbrust, E. V. et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86 (2004)
Ruiz-Trillo, I., Lane, C. E., Archibald, J. M. & Roger, A. J. Insights into the evolutionary origin and genome architecture of the unicellular opisthokonts Capsaspora owczarzaki and Sphaeroforma arctica . J. Eukaryot. Microbiol. 53, 379–384 (2006)
Seo, H. C. et al. Miniature genome in the marine chordate Oikopleura dioica . Science 294, 2506 (2001)
Sullivan, J. C., Reitzel, A. M. & Finnerty, J. R. A high percentage of introns in human genes were present early in animal evolution: evidence from the basal metazoan Nematostella vectensis . Genome Inform. 17, 219–229 (2006)
Halbleib, J. M. & Nelson, W. J. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 20, 3199–3214 (2006)
Gupta, G. & Surolia, A. Collectins: sentinels of innate immunity. Bioessays 29, 452–464 (2007)
Yamaguchi, Y. Lecticans: organizers of the brain extracellular matrix. Cell. Mol. Life Sci. 57, 276–289 (2000)
Zelensky, A. N. & Gready, J. E. The C-type lectin-like domain superfamily. FEBS J. 272, 6179–6217 (2005)
Akiyama, S. K. Integrins in cell adhesion and signaling. Hum. Cell 9, 181–186 (1996)
Erwin, D. H. The origin of metazoan development — a paleobiological perspective. Biol. J. Linn. Soc. 50, 255–274 (1993)
van der Rest, M. & Garrone, R. Collagen family of proteins. FASEB J. 5, 2814–2823 (1991)
Tissir, F. & Goffinet, A. M. Reelin and brain development. Nature Rev. Neurosci. 4, 496–505 (2003)
Suarez-Castillo, E. C. & Garcia-Arraras, J. E. Molecular evolution of the ependymin protein family: a necessary update. BMC Evol. Biol. 7, 23 (2007)
Leadbeater, B. S. Developmental and ultrastructural observations on two stalked marine choanoflagellates, Acanthoecopsis spiculifera Norris and Acanthoeca spectabilis Ellis. Proc. R. Soc. Lond. B 204, 57–66 (1979)
Leadbeater, B. S. C. Developmental studies on the loricate choanoflagellate Stephanoeca diplocostata Ellis. 7. Dynamics of costal strip accumulation and lorica assembly. Eur. J. Protistol. 30, 111–124 (1994)
Hutter, H. et al. Conservation and novelty in the evolution of cell adhesion and extracellular matrix genes. Science 287, 989–994 (2000)
Adamska, M. et al. The evolutionary origin of hedgehog proteins. Curr. Biol. 17, R836–R837 (2007)
Letunic, I. et al. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 34, D257–D260 (2006)
Best, A. A., Morrison, H. G., McArthur, A. G., Sogin, M. L. & Olsen, G. J. Evolution of eukaryotic transcription: insights from the genome of Giardia lamblia . Genome Res. 14, 1537–1547 (2004)
Derelle, R., Lopez, P., Le Guyader, H. & Manuel, M. Homeodomain proteins belong to the ancestral molecular toolkit of eukaryotes. Evol. Dev. 9, 212–219 (2007)
Larroux, C. et al. The NK homeobox gene cluster predates the origin of Hox genes. Curr. Biol. 17, 706–710 (2007)
Knoll, A. H. Life on a Young Planet (Princeton Univ. Press, Princeton, 2003)
Peterson, K. J. & Butterfield, N. J. Origin of the Eumetazoa: testing ecological predictions of molecular clocks against the Proterozoic fossil record. Proc. Natl Acad. Sci. USA 102, 9547–9552 (2005)
Ekman, D., Bjorklund, A. K. & Elofsson, A. Quantification of the elevated rate of domain rearrangements in Metazoa. J. Mol. Biol. 327, 1337–1348 (2007)
Tordai, H., Nagy, A., Farkas, K., Banyai, L. & Patthy, L. Modules, multidomain proteins and organismic complexity. FEBS J. 272, 5064–5078 (2005)
Csuros, M. in Proceedings of the Comparative Genomics: RECOMB 2005 International Workshop; Dublin, Ireland (eds McLysaght, A. & Huson, D. H.) 47–60 (Springer, Berlin, 2005)
Rogozin, I. B., Wolf, Y. I., Sorokin, A. V., Mirkin, B. G. & Koonin, E. V. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 13, 1512–1517 (2003)
Roy, S. W. & Gilbert, W. Complex early genes. Proc. Natl Acad. Sci. USA 102, 1986–1991 (2005)
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004)
Acknowledgements
This work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Livermore National Laboratory, Lawrence Berkeley National Laboratory, and Los Alamos National Laboratory. Work in the King laboratory is supported by funding from the Gordon and Betty Moore Foundation, the Pew Scholars program, and R. Melmon. The Rokhsar group is supported by the Gordon and Betty Moore Foundation and R. Melmon. We thank J. Stajich, P. Johnson and R. Lusk for discussions, E. Hare, E. Meltzer and K. Osoegawa for technical advice, E. Begovic for assistance with Fig. 1, M. Dayel and N. Patel for critical reading of the manuscript, and S. Carroll for early support of this project. N.K. is a Scholar in the Integrated Microbial Biodiversity Program of the Canadian Institute for Advanced Research.
Author Contributions N.K. and D.R. are co-senior authors.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
The file contains Supplementary Figures S1-S12 with Legends, Supplementary Tables S1-S11, Supplementary Notes and additional refrences. (PDF 2819 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
King, N., Westbrook, M., Young, S. et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788 (2008). https://doi.org/10.1038/nature06617
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature06617
This article is cited by
-
Notch intracellular domains form transcriptionally active heterodimeric complexes on sequence-paired sites
Scientific Reports (2024)
-
The polyketide to fatty acid transition in the evolution of animal lipid metabolism
Nature Communications (2024)
-
Origins of cancer: ain’t it just mature cells misbehaving?
The EMBO Journal (2024)
-
What is it like to be a choanoflagellate? Sensation, processing and behavior in the closest unicellular relatives of animals
Animal Cognition (2023)
-
Bioinformatics approaches for classification and investigation of the evolution of the Na/K-ATPase alpha-subunit
BMC Ecology and Evolution (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
