
Abstract
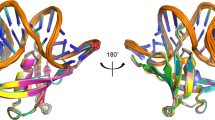
Left-handed Z-DNA is a higher-energy form of the double helix, stabilized by negative supercoiling generated by transcription or unwrapping nucleosomes1. Regions near the transcription start site frequently contain sequence motifs favourable for forming Z-DNA2, and formation of Z-DNA near the promoter region stimulates transcription3,4. Z-DNA is also stabilized by specific protein binding; several proteins have been identified with low nanomolar binding constants5,6,7,8,9. Z-DNA occurs in a dynamic state, forming as a result of physiological processes then relaxing to the right-handed B-DNA1. Each time a DNA segment turns into Z-DNA, two B–Z junctions form. These have been examined extensively10,11,12, but their structure was unknown. Here we describe the structure of a B–Z junction as revealed by X-ray crystallography at 2.6 Å resolution. A 15-base-pair segment of DNA is stabilized at one end in the Z conformation by Z-DNA binding proteins, while the other end remains B-DNA. Continuous stacking of bases between B-DNA and Z-DNA segments is found, with the breaking of one base pair at the junction and extrusion of the bases on each side (Fig. 1). These extruded bases may be sites for DNA modification.

Two bases have been extruded from base stacking at the junction. The white line goes from phosphate to phosphate along the chain. O is shown red, N blue, P yellow and C grey.
Similar content being viewed by others

Main
A 15-base-pair DNA duplex was designed with two overhanging nucleotides at each end. We have taken advantage of the tight binding of the Z-DNA binding domain hZαADAR1 from the human editing enzyme, double-stranded (ds)RNA adenosine deaminase (ADAR1)5,13, to stabilize the Z conformation in one-half of the DNA duplex while the remainder of the DNA duplex retains the B conformation. One B–Z junction is thus formed in the middle of the DNA duplex, connecting the Z- and B-DNA. The hZαADAR1 domain (amino acids 140–202) was co-crystallized with duplex DNA (5′-GTCGCGCGCCATAAACC-3′ and 5′-ACGGTTTATGGCGCGCG-3′). The crystal has space group P61, and the structure of the complex was solved at 2.6 Å resolution (R = 23.8%, Rfree = 28.5%; Supplementary Table 1 and Supplementary Methods).
Two hZαADAR1 peptides bind to each strand of the DNA duplex. Thus, four hZαADAR1 peptides (ZαI–ZαIV) form a complex with one DNA duplex in the crystallographic asymmetric unit (Figs 1 and 2a, and Supplementary Fig. S1). The structures of the four hZαADAR1 domains bound to DNA (ZαI–ZαIV) are nearly identical to that previously described for hZαADAR1 (ref. 5) (Supplementary Fig. S2, Supplementary Table 2 and Supplementary Discussion). The 1:4 stoichiometry between the DNA duplex and hZαADAR1 was confirmed by circular dichroism (CD) measurements (Supplementary Fig. S3). In the DNA duplex, eight base pairs are in the left-handed Z conformation, and six base pairs retain the right-handed B conformation (Figs 1 and 2a). Between the B and Z conformations, an A·T base pair is disrupted and one base from each strand (T0′ and A0) is extruded from the double helix (Figs 1 and 2b; note that base nomenclature is defined in Fig. 2 and its legend). The sugar phosphate backbone of these bases links the B- and Z-DNA segments (Fig. 3); a sharp turn accommodates a reversal in the backbone direction. At the B–Z junction the DNA is bent by 11.05°, and the helical axes are displaced from each other by 5.2 Å, when calculated on the base plane of C - 1 and G1′ (Fig. 3b).

a, Schematic diagram of the DNA sequence and protein positions. One strand has numbers with a prime symbol. Negative numbers start at the 5′ end and increase towards the extruded junctional bases A0 and T0′. Positions of the DNA recognition helix and β-wing of Zα are indicated. There are no contacts between the protein and the extruded bases. b, Stereo 2Fo - Fc omit map of the centre of the molecule with A0 and T0′ omitted, calculated using native data at 2.6 Å. The view is from the rear with respect to Fig. 1.

Red and blue lines connect phosphate groups in Z-DNA and B-DNA, respectively. Numbers refer to bases, as shown in Fig. 2. a, Front view showing A0 (right) and T0′ (left), and the abrupt reversal of helical twist. Distances between stacked bases in the central region (G - 3, C - 2, C - 1, A0, T1, A2 and A3 on one strand, and T - 3′, T - 2′, A - 1′, T0′, G1′, G2′ and C3′ on the other) are indicated. b, Side view with A0 in front. The deviation in helical axes between B- and Z-DNA is shown.
The structure of the eight-base-pair region in the Z conformation is essentially the same as the previously described Z-DNA structure14 (Supplementary Table 3), with a zig-zag sugar phosphate backbone and an alternating anti-syn base conformation. The other end of the DNA duplex adopts a standard B conformation15. No interactions are found between hZαADAR1 and the extruded bases (Fig. 2a), and the glycosyl bonds of A0 and T0′ are both in the anti conformation. The base of T0′ is oriented almost parallel to the helix axis, whereas the A0 base is fully extended out from the helix (Figs 1 and 3, and Supplementary Fig. S1). A segment of an electron density omit map for the extruded thymine T0′ and adenine A0 are shown in Fig. 2b and in Supplementary Fig. S4. The extruded adenine A0 has a much higher temperature factor, consistent with the weak electron density map around adenine A0.
Base-pair parameters and base-pair step parameters of Z- and B-DNA in this structure are consistent with those observed in standard Z- and B-DNA, respectively14,15 (Supplementary Table 3, ref. 16 and Supplementary Discussion, refs 17, 18, 19–20). The single exception is a relatively long rise distance (Dz) of 4.60 Å between the C - 2·G2′ and C - 1·G1′ base pairs in the Z conformation (Fig. 3a and Supplementary Table 3), which may be attributable to the fact that the C - 1·G1′ base pair in the Z conformation has moved from the Z-DNA side to the B-DNA side, in order to stack with T1·A - 1′ in the B conformation (Figs 3 and 4).

Successive changes in the orientation of vectors between deoxyribose atoms C1′ - C1′ of the base pairs is shown, illustrating the left-handed rotation in the upper base pairs and the right-handed reversal in the lower base pair.
The most striking finding in this structure is that the handedness of the DNA duplex can be completely reversed merely by breaking one base pair and extruding the bases from the duplex. Although in this structure an A and a T are extruded, it is equally possible that the extruded bases could be a G and a C. Base stacking is continuous from B-DNA to Z-DNA through the B–Z junction without any abrupt breaks (Figs 1, 3 and 4); this confirms that base stacking constitutes a major stabilizing factor, even within the B–Z junction. Only a slight destabilization of DNA containing B–Z junctions was observed in thermodynamic investigations of the melting of oligomers containing a B–Z junction12; in that study, the transition from B-DNA to the hybrid B/Z DNA structure decreases the free energy of melting by only 0.5 kcal mol-1 relative to the right-handed DNA control. This small energy is not surprising because of the continuity of base stacking through the junction.
The handedness abruptly changes from left to right via a very sharp turn by the phosphate backbone. The junctional angles found between the three phosphate atoms (P - 1, P0 and P1; P - 1′, P0′ and P1′) in the B–Z junction are 91.9° and 59.4°, respectively, while the angles between the three adjacent phosphate atoms of standard B- and of Z-DNA are 150° and 110°, respectively. Some adjustments are found for C - 1 and T1 and their paired bases. Thus, it would be reasonable to consider that the B–Z junction comprises the three base pairs at the -1, 0 and 1 positions. This is consistent with the previous NMR and Raman spectroscopic studies, which asserted that three base pairs or less may be involved in the B–Z junction10,11.
It was apparent in the first Z-DNA structure14 that the conversion from the B to Z conformation occurs with the base pairs ‘flipping over’ and the deoxyguanosine base adopting the syn conformation (Supplementary Fig. S5). Seeing the structure of the B–Z junction inspires the formulation of a model for the B-DNA to Z-DNA transition. Torsional strain causes the breaking of a base pair and extrusion of the bases. The extruded bases are free to flip over and re-form a base pair, thereby nucleating base-pair breaking and extrusion in the adjacent pair of bases. This process could continue, zipper-like, in two directions, to the limit of the Z-DNA forming sequence, with the junctional bases remaining extruded. Additional negative torsional strain in chromatin could lengthen the Z-DNA segment with continuation of the base-pair disruption, extrusion and reformation. At the junctional sites, slight adjustments rearrange base stacking and minimize backbone strain.
It has been established through structural analysis that base extrusion from the double helix is common in base-modifying enzymes21. The similarity of those structures, especially with the conformation of A0, raises the possibility that the extruded bases might also be recognizable as substrates by some base-modifying enzymes. The A0 base is similar in position to that seen in the crystal structures of extruded bases co-crystallized with Hhal methyltransferase22, human DNA repair protein AGT (O(6)-alkylguanine-DNA alkyltransferase)23, or bacteriophage T4 endonuclease V (ref. 24) (Supplementary Fig. S6). In addition, the exposed bases may make them susceptible to chemical modification by oxidizing agents in vivo.
Our knowledge of the biological role of Z-DNA has been broadened by the discovery of four families of proteins that bind to it, most of which have been characterized by protein–Z-DNA crystal structures. These include the editing enzyme ADAR1 (ref. 5), an interferon-inducible Z-DNA binding protein DLM-1 (ref. 6), a pox virus virulence factor protein E3L (refs 7, 8), and more recently an orthologue of the interferon-induced kinase PKR, which has dsRNA binding domains in mammals but contains Z-DNA binding domains in fish (PKZ)9.
There are several observations that point to a role for Z-DNA in transcriptional regulation. The widespread occurrence of sequences favouring Z-DNA formation near the promoter region of genes2 places them in a position where they can trap negative supercoiling, which accumulates behind a moving polymerase or is associated with nucleosome unwrapping. It has been shown that transcription of the colony stimulating factor-1 gene requires Z-DNA formation in that region; and since Z-DNA cannot go into nucleosomes, it leaves a region of DNA free to accept transcription factors3. In other examples, such as the constitutive nucleolin gene, promoter sequences forming Z-DNA down regulate expression25. The most striking biological effect of a Z-DNA binding protein is seen in the vaccinia pox virus interferon resistance protein E3L. Vaccinia virus is lethal for mice, but if the Z-DNA binding ability is weakened, pathogenicity is decreased. If Z-DNA binding is lost, the virus is no longer lethal for the mouse7. Recent experiments have shown that expressing E3L in HeLa cells transactivates some HeLa genes and inhibits apoptosis, both of which are dependent upon Z-DNA binding26.
Every time a section of DNA forms a left-handed Z-DNA segment, two B–Z junctions are formed. The significance of the observed junction is that it requires little structural disruption, preserves helix integrity and, through maximizing base pairing, it minimizes the energetic cost of the junction. This makes it possible to use the Z-DNA conformation more widely and may be related to the widespread occurrence of sequences favouring Z-DNA formation2. B–Z junctions are easily formed in biological systems owing to the widespread occurrence of negative supercoiling. These can be further stabilized by Z-DNA binding proteins. The knowledge that these are associated with extrusion of bases at the junction is likely to open many new avenues of research.
Methods
Crystallization
The protein and DNA duplex in 5 mM HEPES-NaOH pH 7.5 and 10 mM NaCl were mixed to final concentrations of 0.8 mM protein and 0.4 mM DNA (single strand), and crystallized by hanging drop vapour diffusion. Optimized hexagonal crystals were grown in reservoir solutions containing 55–60 mM Na-acetate-HCl pH 4.6, 22–23% MPD and 15–16 mM CaCl2. For selenium MAD analysis, Lys 179 was mutated to methionine.
Structure determinations
A native data set was collected at the BL41-XU beamline of Spring-8 (Harima, Japan). The selenium-labelled crystal data were collected at the BL-6B beamline of PLS (Pohang, Korea) at -80 °C. Diffraction data were processed using HKL200027 and SAINT (Bruker), respectively. Selenium sites were found and refined using SOLVE28 and initial phases were modified using RESOLVE29. The Z-DNA binding protein domains (Zα; chain C, PDB code 1QBJ) were fitted into auto-traced Cα models built by RESOLVE. The Z-DNA model (chains C and D, PDB code 1SFU) and B-DNA model (chains A and B, PDB code 1D23) were fitted into modified electron density maps. Refinements used CNS30 and then Refmac31. The final model contains four hZαADAR1 peptides and one DNA duplex. Data collection phasing and refinement statistics are summarized in Supplementary Table 1.
CD measurement
The CD spectra were obtained at 25 °C, using an AVIV model 202. Protein was gradually added to 5 µM dsDNA in CD buffer (10 mM HEPES, pH 7.4, 10 mM KF and 0.1 mM EDTA), as indicated by N/P ratios (Supplementary Fig. 3). Spectra were recorded at intervals of 1 nm, averaged over 5 s.
For detailed methods, see Supplementary Information.
References
Rich, A. & Zhang, S. Timeline: Z-DNA: the long road to biological function. Nature Rev. Genet. 4, 566–572 (2003)
Champ, P. C., Maurice, S., Vargason, J. M., Camp, T. & Ho, P. S. Distributions of Z-DNA and nuclear factor I in human chromosome 22: a model for coupled transcriptional regulation. Nucleic Acids Res. 32, 6501–6510 (2004)
Liu, R. et al. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106, 309–318 (2001)
Oh, D. B., Kim, Y. G. & Rich, A. Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc. Natl Acad. Sci. USA 99, 16666–16671 (2002)
Schwartz, T., Rould, M. A., Lowenhaupt, K., Herbert, A. & Rich, A. Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 284, 1841–1845 (1999)
Schwartz, T., Behlke, J., Lowenhaupt, K., Heinemann, U. & Rich, A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nature Struct. Biol. 8, 761–765 (2001)
Kim, Y. G. et al. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl Acad. Sci. USA 100, 6974–6979 (2003)
Ha, S. C. et al. A poxvirus protein forms a complex with left-handed Z-DNA: crystal structure of a Yatapoxvirus Zα bound to DNA. Proc. Natl Acad. Sci. USA 101, 14367–14372 (2004)
Rothenburg, S. et al. A PKR-like eukaryotic initiation factor 2a kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl Acad. Sci. USA 102, 1602–1607 (2005)
Sheardy, R. D. & Winkle, S. A. Temperature-dependent CD and NMR studies on a synthetic oligonucleotide containing a B-Z junction at high salt. Biochemistry 28, 720–725 (1989)
Dai, Z. Y., Thomas, G. A., Evertsz, E. & Peticolas, W. L. The length of a junction between the B and Z conformations in DNA is three base pairs or less. Biochemistry 28, 6991–6996 (1989)
Sheardy, R. D., Levine, N., Marotta, S., Suh, D. & Chaires, J. B. A thermodynamic investigation of the melting of B-Z junction forming DNA oligomers. Biochemistry 33, 1385–1391 (1994)
Herbert, A., Lowenhaupt, K., Spitzner, J. & Rich, A. Chicken double-stranded RNA adenosine deaminase has apparent specificity for Z-DNA. Proc. Natl Acad. Sci. USA 92, 7550–7554 (1995)
Wang, A. H. et al. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 282, 680–686 (1979)
Drew, H. R. et al. Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Natl Acad. Sci. USA 78, 2179–2183 (1981)
Lu, X. J., Shakked, Z. & Olson, W. K. A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 300, 819–840 (2000)
Jiang, H., Zacharis, W. & Amirhaeri, S. Potassium permanganate as an in situ probe for B-Z and Z-Z junctions. Nucleic Acids Res. 19, 6943–6948 (1991)
Wolfl, S., Wittig, B. & Rich, A. Identification of transcriptionally induced Z-DNA segments in the human c-myc gene. Biochim. Biophys. Acta 1264, 294–302 (1995)
Mao, C., Sun, W., Shen, Z. & Seeman, N. C. A nanomechanical device based on the B-Z transition of DNA. Nature 397, 144–146 (1991)
Tashiro, R. & Sugiyama, H. A nanothermometer based on the different π stackings of B- and Z-DNA. Agnew. Chem. Int. Edn Engl. 42, 6018–6020 (2003)
Roberts, R. J. & Cheng, X. Base flipping. Annu. Rev. Biochem. 67, 181–198 (1998)
Klimasauskas, S., Kumar, S., Roberts, R. J. & Cheng, X. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76, 357–369 (1994)
Daniels, D. S. et al. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nature Struct. Mol. Biol. 11, 714–720 (2004)
Vassylyev, D. G. et al. Atomic model of a pyrimidine dimer excision repair enzyme complexed with a DNA substrate: structural basis for damaged DNA recognition. Cell 83, 773–782 (1995)
Rothenburg, S., Koch-Nolte, F., Rich, A. & Haag, F. A polymorphic dinucleotide repeat in the rat nucleolin gene forms Z-DNA and inhibits promoter activity. Proc. Natl Acad. Sci. USA 98, 8985–8990 (2001)
Kwon, J.-A. & Rich, A. Biological function of the vaccinia virus Z-DNA–binding protein E3L: Gene transactivation and antiapoptotic activity in HeLa cells. Proc. Natl Acad. Sci. USA 102, 12759–12764 (2005)
Otwinowski, Z. & Minor, W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997)
Terwilliger, T. C. & Berendzen, J. Automated MAD and MIR structure solution. Acta Crystallogr. D 55, 849–861 (1999)
Terwilliger, T. C. Map-likelihood phasing. Acta Crystallogr. D 57, 1763–1775 (2001)
Brunger, A. T. et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 (1998)
Murshudov, G. N., Lebedev, A., Vagin, A. A., Wilson, K. S. & Dodson, E. J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D 55, 247–255 (1999)
Acknowledgements
We thank T. Schwartz for discussions and technical advice. Use of the Pohang Light Source (PLS) was supported by MOST and POSCO. Use of the Spring-8 is supported by the Japan Synchrotron Radiation Research Institute (JASRI). K.K.K was supported by a Basic Science Research Grant from the Korea Research Foundation. A.R. acknowledges support from the National Institutes of Health and the Dana and Ellison Medical Foundations.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Coordinates and structure factor files have been deposited in the Protein Data Bank under accession number 2ACJ. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Supplementary information
Supplementary Discussion
Additional discussion on the results of the study that could not be included in the main text. (PDF 88 kb)
Supplementary Methods
Additional details on the methods used in the study that could not be included in the main text. (PDF 108 kb)
Supplementary Tables
Supplementary Table 1: Data collection phasing and refinement statistics. Supplementary Table 2: Local base pair step parameters of B-Z DNA and standard B- and Z-DNA. Supplementary Table 3 RMS deviation (Å) among Zα domains. (PDF 162 kb)
Supplementary Figure S1
Overall structure of the B-Z junction containing DNA bound to hZαADAR1. (PDF 156 kb)
Supplementary Figure S2
Structural overlap of the Zα proteins bound to Z-DNA. (PDF 193 kb)
Supplementary Figure S3
Titration by CD spectrometry of the 17mer duplex with hZαADAR1. (PDF 124 kb)
Supplementary Figure S4
Stereo 2Fo-Fc omit maps of extruded nucleotides. (PDF 282 kb)
Supplementary Figure S5
A diagram illustrating the B-Z transition and the ensuing conformational changes. (PDF 693 kb)
Supplementary Figure S6
Structures of DNA including extruded bases. (PDF 83 kb)
Rights and permissions
About this article
Cite this article
Ha, S., Lowenhaupt, K., Rich, A. et al. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature 437, 1183–1186 (2005). https://doi.org/10.1038/nature04088
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature04088
This article is cited by
-
The role of ADAR1 through and beyond its editing activity in cancer
Cell Communication and Signaling (2024)
-
Non-canonical DNA structures in the human ribosomal DNA
Histochemistry and Cell Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
