
Abstract
Some people inherit an unfortunate combination of genetic sequences, such that exposure to an external trigger causes their immune response to turn on their own tissues. Although mutations in a single gene can cause autoimmunity, most autoimmune diseases are associated with several sequence variants. Marked advances in genetic resources and tools are now making it possible to identify the sequence variants that contribute to autoimmune diseases — promising a better understanding of how we normally remain tolerant of our own tissue components, and how this goes wrong in autoimmune disease.
Similar content being viewed by others

Main
Autoimmune diseases are major causes of morbidity and mortality throughout the world. Many of these diseases tend to be difficult or impossible to cure, for the obvious reason that the focus of the immune response — self antigens — cannot be eliminated. The physical, psychological and economic burden of these diseases is especially devastating because they often attack young adults. The problem is also compounded by the failure of conventional cellular immunological analyses to shed much light on the pathogenic mechanisms. Recently developed therapies, such as tumour necrosis factor (TNF) antagonists, have had some remarkable successes, but these treatments target resulting organ damage and not the (usually unknown) underlying causes. The realization that the development of autoimmunity is strongly influenced by inherited polymorphisms (or DNA sequence variations) brings hope that understanding the genetics of autoimmune diseases will teach us about the causal derangements, and perhaps lead to new therapeutic strategies. Here, we summarize our current understanding of the genetic basis of autoimmunity. We emphasize principles, rather than attempt to list all the reported disease-associated polymorphisms. A brief introduction to the mechanisms of self tolerance and its breakdown provide the foundation for the subsequent discussion of the genetics of autoimmune diseases.
Self tolerance and autoimmunity
All individuals are tolerant of their own potentially antigenic substances, and failure of self tolerance is the fundamental cause of autoimmunity. The mechanisms of self tolerance have been worked out in considerable detail in animal models, and are best understood for CD4+ T cells. Self tolerance can be divided into central tolerance and peripheral tolerance. In central tolerance, immature lymphocytes that happen to recognize self antigens in generative lymphoid organs (the bone marrow for B cells and the thymus for T cells) die by apoptosis; in peripheral tolerance, mature self-reactive lymphocytes encounter self antigens in peripheral tissues and are killed or shut off. The principal mechanisms of peripheral tolerance are anergy (functional unresponsiveness), deletion (apoptotic cell death), and suppression by regulatory T cells1. These mechanisms are described in more detail in the reviews in this issue by Goodnow et al. (page 590), and by Kronenberg and Rudensky (page 598). Autoimmune diseases develop when self-reactive lymphocytes escape from tolerance and are activated. Although the mechanisms by which this occurs are not entirely known, autoimmunity is thought to result from a combination of genetic variants, acquired environmental triggers such as infections, and stochastic events.
Genetics of single-gene disorders
To assess the contribution of genetic factors to disease susceptibility, genetic epidemiologists examine the extent of familial clustering; the degree to which monozygotic twins are more concordant for the presence of a disease compared with dizygotic twins; and the increased risk that family members of persons with disease will develop that disease compared with an individual from the general population. Using such estimates of genetic risk, it becomes obvious that in single-gene disorders, the risk conferred on an individual by a given genetic variant is very high, but the overall impact on the population is minimal because these variants are rare.
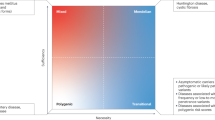
In these ‘simple’ diseases (or traits), although there is often variation in phenotypic expression, the relationship between the causal genetic variant and the disease state is deterministic (Fig. 1a). Genome-wide linkage studies attempt to identify genetic markers (and thus a genomic region) where there is more sharing of alleles between individuals (with a given trait) within families than is statistically expected. Such studies are most commonly used in identifying causal genetic variants for single-gene disorders. Genetic studies of simple traits undoubtedly met with earlier success than those of complex traits. However, recent advances in the study of complex traits are now contributing to this knowledge.

a, In simple mendelian traits, the relationship between the causal genetic variant (genotype) and the disease state is deterministic. b, In complex traits, the clinically recognized disease state results from interactions between multiple genotypes and the environment. Individual genotypes can affect one or more components of the adaptive or innate immune systems; together these lead to an altered immune response to self antigens. On the basis of current findings, the influence of any individual causal allele is modest, and therefore the relationship between the causal variant and the disease state is probabilistic. Although still providing an incomplete picture, the genetic discoveries in mendelian and common diseases are beginning to help build a model of autoimmune disease. The ultimate goal is to build a specific model for each individual disease whereby the effect of individual risk factors (genetic and non-genetic), their interactions, and their impact on disease susceptibility, disease progression and clinical management, are understood.
Since inborn errors of metabolism were identified as diseases caused by mutations or deletions affecting single genes, clinicians and scientists have realized that simple genetic traits can teach us a great deal about the pathways of disease, and about normal physiology. In some cases, the genes identified in simple disorders will have more subtle alterations that confer susceptibility to a ‘common’ disease (that is, diseases caused by a combination of alleles); in other cases, the mutations will simply suggest which biological pathways may be implicated in common diseases. Although many examples exist of single-gene knockout experimental models that have led to autoimmunity, here we focus on genes that are known to be involved in human disorders (Table 1).
AIRE and central tolerance
AIRE (autoimmune regulator) was identified as the gene that is mutated in autoimmune polyendocrine syndrome (APS-1) — a disorder that manifests as autoimmune attack against multiple endocrine organs, the skin and other tissues2. In a tour de force of functional genomics, the mouse homologue of the gene has been knocked out, and the AIRE protein shown to be responsible for the thymic expression of some antigens that are expressed at high levels in different peripheral tissues. In the absence of thymic expression, T cells specific for these antigens escape negative selection (central tolerance), enter the periphery and attack the target tissues3,4. The possibility that thymic defects underlie autoimmunity was raised long before this elegant demonstration of the importance of AIRE-dependent thymic selection in self tolerance. For example, one of the susceptibility genes associated with type 1 diabetes in humans is insulin: it has been suggested that the disease-associated polymorphism of the insulin gene reduces its expression in the thymus, thus allowing insulin-reactive T cells to escape deletion5. It remains to be determined whether defects in central tolerance contribute significantly to most multigenic autoimmune diseases6.
CTLA4 and T-cell anergy
Cytotoxic T lymphocyte antigen 4 (CTLA4; CD152) is an inhibitory receptor expressed by T cells that recognizes the costimulatory molecules B7-1 (CD80) and B7-2 (CD86), the ligation of which shuts off T-cell responses and promotes long-lived anergy7. CTLA4 works by competitively blocking the engagement of the activating receptor CD28 (by CD80 or CD86), and by transducing inhibitory signals; the latter probably involves tyrosine and serine/threonine phosphatase activation8. Germline CTLA4 knockout mice develop a fatal syndrome of multi-organ lymphocytic infiltrates and severe enlargement of lymphoid organs. Such symptoms are consistent with a systemic autoimmune reaction presumably directed against multiple, as yet unknown, self antigens. This marked demonstration of the obligatory function of CTLA4 has led to a search for polymorphisms in the CTLA4 gene that are associated with autoimmune diseases. A surprising discovery was that several such diseases, including Graves' disease, type 1 diabetes and other endocrinopathies, show a striking association with a CTLA4 polymorphism that results in reduced production of a truncated splice variant, which has inhibitory activity9. The functional consequences of producing this altered form of CTLA4 have not been defined, but as expected for a complex disease, the biological effect of the causal allele is more subtle than what is found in monogenic disorders or in knockout mice.
FOXP3 and regulatory T cells
FOXP3 (encoding a transcription factor of the forkhead family) is a striking example of a gene whose role in autoimmunity has been revealed by the confluence of animal studies and studies of a quite rare human disease. CD4+CD25+ regulatory T cells, now established as major controllers of immune responses to self and other antigens10, were shown to express high levels of FOXP3. Three groups demonstrated that induced knockout or spontaneous mutation of the mouse Foxp3 gene (‘scurfy’ mice) led to a systemic autoimmune disease associated with the absence of CD4+CD25+ regulatory T cells11,12,13. At the same time, a human disease known by the acronym IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome) was shown to be associated with mutations in FOXP3. These results indicate that the development and/or function of regulatory T cells are dependent on the activity of this one transcription factor; its downstream targets and major functions are not yet defined.
FAS and lymphocyte apoptosis
Fas (CD95) is the prototype of a death receptor of the TNF receptor family. Its biological importance was established by seminal studies demonstrating that in two mouse models of autoimmunity and lymphoproliferation (named the lpr/lpr and gld/gld strains), genetic lesions affected the genes encoding Fas and Fas ligand, respectively14. These results were among the first to show that a single genetic abnormality could give rise to a complex autoimmune phenotype, and that the failure of one apoptosis pathway resulted in self-tolerance breakdown. The Fas death receptor contributes to the deletion of mature T and B cells that recognize self antigens15. A human disease, called autoimmune lymphoproliferative syndrome (ALPS), which resembles the disease of lpr and gld mice, is caused by Fas mutations16. Thus, this is an excellent example of how mouse models can lead to a better understanding of human diseases. Although these autoimmune diseases have a superficial similarity to human systemic lupus erythematosus (SLE), there is no convincing evidence for FAS polymorphisms or mutations being associated with SLE.
The elegant simplicity of these monogenic disorders has been key to using them to elucidate mechanisms of self tolerance and autoimmunity. The experimental models also illustrate how animal studies inform analyses of human disease, and the striking influences of ‘background’ genes on the severity and manifestations of diseases. However, as stated at the outset, these single-gene models of autoimmunity are rare; most autoimmune diseases are complex, multigenic traits. The remainder of our discussion will focus on these complex disorders.
Genetics of common autoimmune diseases
In contrast to simple diseases, common diseases are believed to result from a combination of susceptibility alleles at multiple loci, environmental factors (such as smoking, pathogen exposure and hormone levels), and stochastic events (Fig. 1b). There is some debate as to whether common diseases are caused by common alleles of low penetrance (that is, alleles that confer modest increased risk to disease) or by multiple rare alleles of high penetrance. Although most disease alleles identified for common autoimmune diseases have the former characteristics (Table 2), these may not represent the full spectrum of disease alleles. Our ability to discover as yet unidentified disease alleles has been improved by the recent explosion of knowledge regarding the common genetic variation in the human genome. Specifically, recent work has demonstrated that the vast majority of genetic variation in the human genome consists of individual bases that exist as either of two alleles in the population — known as single nucleotide polymorphisms (SNPs) — rather than as deletions or rearrangements. Approximately ten million SNPs in the human genome have a minor allele frequency greater than 1% and represent about 90% of the genetic variation in the human genome. Initial efforts to discover and map SNPs to the reference sequence of the human genome have resulted in a public resource containing most of these common SNPs17,18,19.
Over the past decade, in autoimmunity as in many other human diseases, there has been great interest in testing candidate genomic regions (identified by linkage studies), or candidate genes (selected on the basis of their location under a linkage peak, or their known functional properties, or both) for evidence of their association with disease. Before the creation of public SNP databases, sequencing was needed to identify the genetic variants to test in an association study. Now it seems that there is almost an excess of SNPs to type: do we need to type all ten million to find the few that are causing a given autoimmune disease? To answer this question, it is important to understand the relationships that exist between these SNPs. First, by examining a high density of specific areas of the genome20,21,22, and by performing genome-wide surveys23, it has become clear that the alleles of the SNPs form patterns (also known as haplotypes) in the genome. Specifically, the emerging data demonstrate that alleles at nearby SNPs are highly correlated with one another (this is known as linkage disequilibrium). The tight correlation between SNPs, however, is broken down by recombination events resulting in haplotype blocks. Furthermore, the existing data and models based on these data suggest that, rather than occurring randomly, there are recombination hotspots in the genome22,24,25. Knowledge of the haplotype structure not only allows an optimal subset of SNPs to be selected that efficiently extracts the information about the common patterns of variation, but it also can direct how such data should be analysed. For these reasons, an international effort is underway to map the common patterns of genetic variation across the entire genome, and it is expected that the first phase will be completed by mid-2005 (ref. 26). Already the information from this International HapMap can be incorporated into the design of association studies of candidate genes/genomic regions, and it will soon be possible to apply this new information to the design and execution of powerful genome-wide association studies27,28.
Successes in mapping susceptibility genes
Given the early lack of success in identifying genes for complex traits, it was believed that successes in human disease genetics might be limited to disorders with single-gene mendelian inheritance. However, this changed with the publication of several studies reporting the mapping of susceptibility loci in complex human traits: many of these studies took advantage of the haplotype structure of the genome to narrow down the region of association. Early in 2000, fine-scale mapping of a complex human trait successfully led to the localization of the type 1 diabetes locus in the major histocompatibility complex (MHC) to a discrete 570-kilobase (kb) region29. An association mapping approach also proved successful when Hugot and colleagues identified sequence variants in the NOD2 (CARD15) gene that are associated with Crohn's disease30. Comprehensive association mapping approaches successfully identified haplotypic variation in the cytokine gene cluster on chromosome 5q31, which confers susceptibility to Crohn's disease31, for which two potentially causal variants have recently been proposed32. The ADAM33 and GPRA susceptibility genes for asthma33 have similarly been identified. Large association studies of candidate genes have also resulted in important discoveries: the IDDM12/CTLA4 locus in Graves' disease and in type 1 diabetes9; the NOD2 gene in Crohn's disease34,35; the PTPN22 gene in type 1 diabetes, rheumatoid arthritis and SLE36,37,38,39; and the PDCD1 gene in SLE and rheumatoid arthritis40,41. The observation that genes such as CTLA4 and PTPN22 are associated with multiple disorders is consistent with the hypothesis that certain immunological pathways are common to multiple autoimmune diseases, whereas other pathophysiological mechanisms are specific to a particular disease.
The identification of these susceptibility genes provides the immunology community with an opportunity to study the key pathways and molecular mechanisms that can lead to increased disease susceptibility. Initial functional studies have begun to provide some relevant clues. For example, work regarding the effect of genetic variants in CTLA4 on susceptibility to type 1 diabetes in mice and humans has provided substantial evidence that modulating the expression of alternative splice forms can regulate the expression of T-cell-mediated autoimmunity9,42. Specifically, the human variant leads to the decreased production of a ligand-independent form of CTLA4 — a splice form believed to be involved in the downregulation of memory/effector T-cell activation9,42. Genetic variation in the human PTPN22 gene also seems to modulate T-cell activity. However, this is due to an amino acid change in the encoded protein, the lymphoid protein tyrosine phosphatase (LYP), a known suppressor of T-cell activation. Bottini and colleagues43 hypothesized that PTPN22 was a good candidate gene for autoimmunity because it and other protein tyrosine phosphatases are involved in preventing spontaneous T-cell activation. These investigators identified an SNP that was associated with type 1 diabetes43, which changed an amino acid involved in the interaction between LYP and the negative regulatory kinase CSK. PDCD1, a positional and functional candidate gene, encodes the programmed cell death 1 gene whose gene product has been shown to regulate peripheral tolerance in T and B cells44,45. An intronic variant in the PDCD1 gene was proposed to lead to aberrant PDCD1 regulation and increase an individual's susceptibility to SLE41. These few examples demonstrate how variants that modify gene expression or protein structure can lead to modulation of the adaptive immune responses.
Alteration of the innate immune system, on the other hand, may be the primary effect of the two missense mutations and the truncation mutant in the NOD2 gene, all of which have been associated with Crohn's disease. Specifically, the NOD2 gene product has been shown to recognize muramyl dipeptide (MDP) — a component of the bacterial wall. The Crohn's disease-associated mutations were believed to lead to impaired nuclear factor-κB (NF-κB) signalling following recognition of MDP34,46,47. Using spleen macrophages derived from Nod2-deficient mice, Watanabe and colleagues proposed an alternative explanation. Specifically, they suggested that the NOD2 gene product limits the pro-inflammatory effects mediated by Toll-like receptor 2 (TLR2) signalling, such that mutations in NOD2 would lead to excessive T-helper type 1 (TH1) responses46,47. Efforts to knock out the Nod2 gene have not, however, led to gastrointestinal inflammation. This perhaps is not surprising given the complexity of the human disease; no single genetic or environmental factor is expected to be necessary or sufficient to cause the disease. These Nod2−/− mice also did not show differential susceptibility to chemically induced gastrointestinal inflammation, but they did seem to be more susceptible to oral challenge with Gram-positive bacteria48,49. The results from knocking-in the human frameshift mutation into mice (Nod22939iC ), however, seem to suggest a very different mechanism: as opposed to the lack of response to MDP in the Nod2−/− mice, the Nod22939iC mice have an elevated response to MDP and a heightened intestinal inflammatory response to chemical injury50.
These exciting experiments demonstrate the importance of a genetic and environmental context for such functional studies and the challenges that lie ahead in deciphering the exact mechanisms by which genetic variation can lead to autoimmunity. Although it is still too early to know the precise mechanisms by which individual allelic variants confer susceptibility, the elucidation of the pathogenic mechanisms in the coming years will undoubtedly shed much light on the common and disease-specific processes.
Challenges in mapping susceptibility genes
Genetic association studies of autoimmune diseases, as for all other complex human traits, face two main challenges: the ability to distinguish between true and false associations, and the ability to demonstrate causality. The first challenge relates to the nature of many causal alleles in common disease; many of the implicated alleles only confer a modest increased risk of developing disease (see Table 2). The power to detect disease susceptibility genes is influenced by the magnitude of the risk conferred by the susceptibility allele, and by its frequency in the population. The weaker the effect of an allele, the greater the sample size required to detect an association signal that can be distinguished from the background noise of an association study. Unfortunately, it is not possible to know a priori the frequency and the strength of disease alleles. However, the detection of modest alleles may require many thousands of samples28. This is further complicated by the fact that the first report of an association often overestimates the effect of the putative associated allele51. This effect is known as the ‘winner's curse’ — a mathematical concept first described in the context of auctions and bilateral negotiations52. Replication studies, therefore, often produce seemingly inconsistent results when compared with the original study53,54. Luckily, this challenge can be addressed by performing association studies (novel and replication) with large sample sizes, and by combining the results from multiple studies using a meta-analysis approach53,55. Indeed, these two approaches have successfully demonstrated that a number of susceptibility genes for autoimmune disease (for example, PTPN22 in type 1 diabetes, IBD5 and NOD2 in Crohn's disease) can be convincingly identified and replicated37,56,57.
The second challenge relates to the patterns of linkage disequilibrium described above. Although intervals of historical recombination occur between haplotype blocks, this is a modest enough effect for correlations between blocks to persist21. Given the extreme local variation in the magnitude of this recombination25 and in gene density19, genomic regions implicated by association studies may contain a single gene or multiple genes. For the latter, current sample sizes may be insufficient to narrow down the implicated region to less than a handful of genes. One example is a 250-kb haplotype on chromosome 5q31 that was found to be associated with Crohn's disease31. The haplotype associated with increased risk of developing Crohn's disease extends uninterrupted across multiple blocks, as the recombination events that have occurred in the intervals in this region have primarily involved the haplotypes that do not confer risk. Therefore, this association implicates a region that contains a minimum of four genes (IRF1, SLC22A5 (OCTN2), SLC22A4 (OCTN1) and PDLIM3) and multiple SNPs that have an allele that is unique to the risk haplotype. Therefore, the identification of the causal gene(s) and related variant(s) will rely on functional studies that link gene/variants and phenotype. Another important example is that of the MHC, where extended haplotypes are believed to represent a common feature of this region58. Importantly, the MHC region has been associated with almost all autoimmune diseases and contains the HLA genes: HLA gene products are crucial for antigen presentation to cells of the adaptive immune system and for controlling some reactions of the innate immune system, such as activation of natural killer (NK) cells. This genomic region also contains hundreds of other genes, many with putative or proven function in the immune system59. Most association studies of this genomic region, however, have been limited to one or a few of the HLA genes. The preliminary efforts to map the patterns of genetic variation in this region suggest that dense sets of SNPs applied to large study cohorts will enable the identification of the HLA and non-HLA components of the genetic susceptibility conferred by the MHC region60,61,62,63.
Future prospects
One of the great promises of genetics is to help build a molecular model of disease that combines information regarding genotype and environment, as well as their various interactions (Fig. 1b). One challenge that is specific to immune-related disorders is the fact that an individual's functional repertoire of specific antigen receptors (B-cell receptors and T-cell receptors) is a product of the interaction between their genetic repertoire and the environment. It will therefore be important to define an individual's functional state in the context of their genetic background. Despite this challenge, some of the discoveries so far in single- gene and complex forms of autoimmunity are helping to build such a model. These models could potentially help to assess an individual's risk of developing disease. There is also some indication that this information will be relevant for assigning patients to molecular subgroups of these very heterogeneous diseases, which may help to predict a particular disease outcome and/or response to therapy.
A complementary approach to improving the response to current therapies is to examine variation in the genes controlling the absorption, distribution, metabolism and excretion of the drugs used to dampen the chronic inflammatory response in patients64. An aspect of these models that may prove to be important is knowing which genes are common to more than one autoimmune disease versus those that are disease-specific, as these might turn out to be qualitatively very different drug targets. Finally, there is no doubt that any novel susceptibility gene, or even newly identified causal variation in a previously well characterized gene, will provide the immunologist with some very interesting avenues to follow in their path to understanding the fundamental mechanisms of autoimmunity.
References
Walker, L. S. & Abbas, A. K. The enemy within: keeping self-reactive T cells at bay in the periphery. Nature Rev. Immunol. 2, 11–19 (2002).
Bjorses, P., Aaltonen, J., Horelli-Kuitunen, N., Yaspo, M. L. & Peltonen, L. Gene defect behind APECED: a new clue to autoimmunity. Hum. Mol. Genet. 7, 1547–1553 (1998).
Anderson, M. S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002).
Liston, A., Lesage, S., Wilson, J., Peltonen, L. & Goodnow, C. C. Aire regulates negative selection of organ-specific T cells. Nature Immunol. 4, 350–354 (2003).
Eisenbarth, G. S. Insulin autoimmunity: immunogenetics/immunopathogenesis of type 1A diabetes. Ann. NY Acad. Sci. 1005, 109–118 (2003).
Mathis, D. & Benoist, C. Back to central tolerance. Immunity 20, 509–516 (2004).
Salomon, B. & Bluestone, J. A. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 19, 225–252 (2001).
Baroja, M. L. et al. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 168, 5070–5078 (2002).
Ueda, H. et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511 (2003).
Sakaguchi, S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22, 531–562 (2004).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 4, 330–336 (2003).
Khattri, R., Cox, T., Yasayko, S. A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature Immunol. 4, 337–342 (2003).
Nagata, S. & Suda, T. Fas and Fas ligand: lpr and gld mutations. Immunol. Today 16, 39–43 (1995).
Lenardo, M. et al. Mature T lymphocyte apoptosis-immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 17, 221–253 (1999).
Fischer, A., Rieux-Laucat, F. & Le Deist, F. Autoimmune lymphoproliferative syndromes (ALPS): models for the study of peripheral tolerance. Rev. Immunogenet. 2, 52–60 (2000).
Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001).
Wang, D. G. et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 280, 1077–1082 (1998).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Patil, N. et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294, 1719–1723 (2001).
Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J. & Lander, E. S. High-resolution haplotype structure in the human genome. Nature Genet. 29, 229–232 (2001).
Jeffreys, A. J., Kauppi, L. & Neumann, R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nature Genet. 29, 217–222 (2001).
Gabriel, S. B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Reich, D. E. et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nature Genet. 32, 135–142 (2002).
McVean, G. A. et al. The fine-scale structure of recombination rate variation in the human genome. Science 304, 581–584 (2004).
The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
Hirschhorn, J. N. & Daly, M. J. Genome-wide association studies for common diseases and complex traits. Nature Rev. Genet. 6, 95–108 (2005).
Wang, W. Y., Barratt, B. J., Clayton, D. G. & Todd, J. A. Genome-wide association studies: theoretical and practical concerns. Nature Rev. Genet. 6, 109–118 (2005).
Herr, M. et al. Evaluation of fine mapping strategies for a multifactorial disease locus: systematic linkage and association analysis of IDDM1 in the HLA region on chromosome 6p21. Hum. Mol. Genet. 9, 1291–1301 (2000).
Hugot, J. P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411, 599–603 (2001).
Rioux, J. D. et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nature Genet. 29, 223–228 (2001).
Peltekova, V. D. et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nature Genet. 36, 471–475 (2004).
Laitinen, T. et al. Characterization of a common susceptibility locus for asthma-related traits. Science 304, 300–304 (2004).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606 (2001).
Hampe, J. et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet 357, 1925–1928 (2001).
Onengut-Gumuscu, S., Ewens, K. G., Spielman, R. S. & Concannon, P. A functional polymorphism (1858C/T) in the PTPN22 gene is linked and associated with type I diabetes in multiplex families. Genes Immun. 5, 678–680 (2004).
Smyth, D. et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53, 3020–3023 (2004).
Kyogoku, C. et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am. J. Hum. Genet. 75, 504–507 (2004).
Begovich, A. B. et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 75, 330–337 (2004).
Lin, S. C. et al. Association of a programmed death 1 gene polymorphism with the development of rheumatoid arthritis, but not systemic lupus erythematosus. Arthritis Rheum. 50, 770–775 (2004).
Prokunina, L. et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nature Genet. 32, 666–669 (2002).
Vijayakrishnan, L. et al. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 20, 563–575 (2004).
Bottini, N. et al. A functional variant of lymphoid tyrosine phosphatase is associated with type 1 diabetes. Nature Genet. 36, 337–338 (2004).
Nishimura, H. & Honjo, T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 22, 265–268 (2001).
Blank, C. et al. Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J. Immunol. 171, 4574–4581 (2003).
Watanabe, T., Kitani, A., Murray, P. J. & Strober, W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nature Immunol. 5, 800–808 (2004).
Girardin, S. E. et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872 (2003).
Pauleau, A. L. & Murray, P. J. Role of Nod2 in the response of macrophages to Toll-like receptor agonists. Mol. Cell. Biol. 23, 7531–7539 (2003).
Kobayashi, K. S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Maeda, S. et al. Nod2 mutation in Crohn's disease potentiates NF-κB activity and IL-1β processing. Science 307, 734–738 (2005).
Ioannidis, J. P., Ntzani, E. E., Trikalinos, T. A. & Contopoulos-Ioannidis, D. G. Replication validity of genetic association studies. Nature Genet. 29, 306–309 (2001).
Bazerman, M. & Samuelson, W. The Winners Curse — an Empirical Investigation. Lecture Notes in Economics and Mathematical Systems 213, 186–200 (1983).
Lohmueller, K. E., Pearce, C. L., Pike, M., Lander, E. S. & Hirschhorn, J. N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nature Genet. 33, 177–182 (2003).
Hirschhorn, J. N., Lohmueller, K., Byrne, E. & Hirschhorn, K. A comprehensive review of genetic association studies. Genet. Med. 4, 45–61 (2002).
Ioannidis, J. P., Trikalinos, T. A., Ntzani, E. E. & Contopoulos-Ioannidis, D. G. Genetic associations in large versus small studies: an empirical assessment. Lancet 361, 567–571 (2003).
Daly, M. J. & Rioux, J. D. New approaches to gene hunting in IBD. Inflamm. Bowel Dis. 10, 312–317 (2004).
Hugot, J. P. Genetic origin of IBD. Inflamm. Bowel Dis. 10, (Suppl. 1) S11–S15 (2004).
Alper, C. A., Awdeh, Z. & Yunis, E. J. Conserved, extended MHC haplotypes. Exp. Clin. Immunogenet. 9, 58–71 (1992).
Horton, R. et al. Gene map of the extended human MHC. Nature Rev. Genet. 5, 889–899 (2004).
Stenzel, A. et al. Patterns of linkage disequilibrium in the MHC region on human chromosome 6p. Hum. Genet. 114, 377–385 (2004).
Walsh, E. C. et al. An integrated haplotype map of the human major histocompatibility complex. Am. J. Hum. Genet. 73, 580–590 (2003).
Ahmad, T. et al. Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum. Mol. Genet. 12, 647–656 (2003).
Miretti, M. M. et al. A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 76, 634–646 (2005).
Ahmadi, K. R. et al. A single-nucleotide polymorphism tagging set for human drug metabolism and transport. Nature Genet. 37, 84–89 (2005).
Tivol, E. A. et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995).
Waterhouse, P. et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4 . Science 270, 985–988 (1995).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3 . Nature Genet. 27, 20–21 (2001).
Fisher, G. H. et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81, 935–946 (1995).
Atkinson, J. P. Complement deficiency: predisposing factor to autoimmune syndromes. Clin. Exp. Rheumatol. 7, (Suppl. 3) S95–S101 (1989).
Acknowledgements
We thank J. Bluestone, M. Anderson, M. Daly and F. Sanchez, for their helpful comments. We also thank L. Gaffney for her help in the preparation of the manuscript. J.D.R. is supported by grants from the CCFA and NIDDK.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Rioux, J., Abbas, A. Paths to understanding the genetic basis of autoimmune disease. Nature 435, 584–589 (2005). https://doi.org/10.1038/nature03723
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature03723
This article is cited by
-
DLA class II risk haplotypes for autoimmune diseases in the bearded collie offer insight to autoimmunity signatures across dog breeds
Canine Genetics and Epidemiology (2019)
-
Deubiquitylating enzymes and drug discovery: emerging opportunities
Nature Reviews Drug Discovery (2018)
-
Structural Evaluation and Binding Mode Analysis of CCL19 and CCR7 Proteins—Identification of Novel Leads for Rheumatic and Autoimmune Diseases: An Insilico study
Interdisciplinary Sciences: Computational Life Sciences (2018)
-
Autoimmunity in plants
Planta (2018)
-
Update on mesenchymal stem cell-based therapy in lupus and scleroderma
Arthritis Research & Therapy (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
