
Abstract
Selective pressures on proteins are usually measured by comparing nucleotide sequences1. Here we introduce a method to detect selection on the basis of a single genome sequence. We catalogue the relative strength of selection on each gene in the entire genomes of Mycobacterium tuberculosis and Plasmodium falciparum. Our analysis confirms that most antigens are under strong selection for amino-acid substitutions, particularly the PE/PPE family2 of putative surface proteins in M. tuberculosis and the EMP1 family3 of cytoadhering surface proteins in P. falciparum. We also identify many uncharacterized proteins that are under strong selection in each pathogen. We provide a genome-wide analysis of natural selection acting on different stages of an organism's life cycle: genes expressed in the ring stage4 of P. falciparum are under stronger positive selection than those expressed in other stages of the parasite's life cycle. Our method of estimating selective pressures requires far fewer data than comparative sequence analysis, and it measures selection across an entire genome; the method can readily be applied to a large range of sequenced organisms.
Similar content being viewed by others


Main
Historically, the study of natural selection has been pursued under a comparative paradigm5. Genes under selection are identified by comparing homologous nucleotide sequences sampled either from different individuals within a species (for example, nucleotide polymorphism studies) or from different species (for example, phylogenetic analysis). The widely used non-synonymous to synonymous substitution ratio (dN/dS) falls within this model6, as do all other existing methods for detecting natural selection on coding sequences7,8,9. Within the comparative paradigm, it would be impossible to measure selective pressures on the basis of a genome sequence from a single individual.
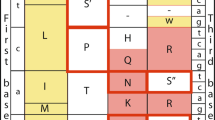
Here we present a method for rapidly detecting differential selective pressures on genes by inspecting a single genome sequence for a footprint of non-synonymous substitutions. Our method rests on a simple observation: if a protein coding region of a nucleotide sequence has undergone an excess number of amino-acid substitutions, then the region will on average contain an overabundance of ‘volatile’ codons, compared with the genome as a whole. For each of the 61 sense codons, we define its volatility as the proportion of its point-mutation neighbours that encode different amino acids (see Fig. 1). The volatility of a codon will be used to quantify the chance that the most recent nucleotide mutation to that codon caused an amino-acid substitution.

The volatility of each codon depends only on the structure the genetic code. The codon CGA, encoding arginine, has eight potential ancestor codons; that is, non-stop codons that differ from CGA by one point mutation. Four of the potential ancestor codons of CGA encode an amino acid different from arginine. Thus the volatility of CGA equals 4/8. The codon AGA also encodes arginine but has a volatility of 6/8. There are 22 codons that have at least one synonym with a different volatility. We use volatility to quantify the chance that the most recent accepted mutation to a site caused an amino-acid change. Letters in parentheses are one-letter amino-acid codes.
Using the concept of codon volatility, we can scan an entire genome to find genes that show significantly more, or less, pressure for amino-acid substitutions than the genome as a whole. If a gene contains many residues under pressure for amino-acid replacements, then the resulting codons in that gene will on average exhibit elevated volatility, because its ancestor codons encoded different amino acids from those encoded by the current codons. Similarly, if a gene is under strong purifying selection not to change its amino acids, then the resulting sequence will on average exhibit lower volatility10.
We assess the statistical significance of each gene's observed volatility by comparing it with a bootstrap distribution of alternative synonymous sequences, drawn according to the background codon usage in the genome (see Methods). This randomization procedure controls for the gene's length and amino-acid composition. As a result of this procedure we obtain a two-sided ‘volatility P value’ for each gene, indicating whether the gene is more, or less, volatile than the genome as a whole. A P value near zero indicates significantly elevated volatility, whereas a P value near one indicates significantly depressed volatility.
Our method of estimating selective pressures by using volatility does not assume a constant mutation rate across sites. If a particular gene experiences a higher nucleotide mutation rate, the gene's volatility will not be biased. Volatility simply measures the chance that the most recent accepted nucleotide mutation in the series of mutations that gave rise to the observed current codon caused a change in the amino acid that is currently encoded. The timing of the most recent accepted mutation can vary from site to site; nevertheless, regardless of its timing, the probability that a site's most recent substitution caused a non-synonymous change is greater (smaller) for a site under positive (negative) selection.
Using the method of codon volatility, we have estimated selective pressures across the complete genomes of M. tuberculosis strain CDC1551 (4,099 unambiguous coding sequences) and of P. falciparum strain Pf3D7 (5,440 unambiguous coding sequences). Table 1 summarizes the volatility P values for the most volatile genes in each genome. The P values for all genes are reported in the Supplementary tables. Each pathogen exhibits a substantial proportion of genes that show signs of much stronger pressure for amino-acid substitutions than the genome as a whole (Fig. 2). We also find a substantial proportion of genes that show much stronger purifying selection than the genome as a whole (Fig. 2). For both pathogens, the genes with extreme volatility are distributed throughout the genome.

In the absence of differential evolutionary pressures between genes—and, in particular under completely neutral evolution—the distribution of P values would be uniform. Instead, each genome shows a characteristic U-shaped distribution, with a significant (Kolmogorov–Smirnov, P < 10-6) excess number of genes with low or high P values. The non-random tails of these distributions indicate a large number of genes under greater pressure for amino-acid substitutions than the genome as a whole, and a large number of genes under greater pressure against amino-acid substitutions.
The PE and PPE gene families of M. tuberculosis, which are putatively expressed on the extracellular surface and exhibit extensive non-synonymous variability2, have been identified as potential antigens for the host immune response11,12,13. The PE family (81 genes) and the PPE family (57 genes) both exhibit significantly greater volatility than the other genes in the M. tuberculosis genome (Pw = 6 × 10-8 for PE and Pw = 3 × 10-22 for PPE; Wilcoxon test). In fact, the PE and PPE genes are ten times more frequent among the 100 most volatile M. tuberculosis genes than they are in the genome as a whole. The elevated volatility of these genes indicates increased pressure for amino-acid substitutions, presumably because of diversifying selection mediated by interactions with the host immune system12,13. These results, based on a single genome sequence, agree with an extensive study in which the authors compared the genomes of two fully sequenced M. tuberculosis strains2. In that study, the authors found the PE/PPE gene families to have the highest non-synonymous to synonymous substitution ratio (dN/dS) among all families with a significantly elevated substitution rate2.
A recent, genome-wide mutagenesis study of M. tuberculosis identified 614 genes essential for optimal growth of the bacterium14. These genes are highly conserved among related organisms and are presumably under stronger purifying selection than the remaining non-essential genes14. The 614 essential genes are significantly less volatile than the non-essential genes (Pw = 4 × 10-6), confirming that the volatility method correctly detects purifying selection. Those genes of M. tuberculosis that are both essential and exhibit low volatility are excellent candidates for drug targets, because their disruption is lethal and their sequences are more stable than those of other genes.
Although the genetic, cellular, life-history and population structures of the eukaryote P. falciparum are more complicated than those of the bacterium M. tuberculosis, an analysis by codon volatility produces a similarly detailed and biologically reasonable picture of differential selection across the P. falciparum genome. The 49 P. falciparum genes denoted as antigens—including asparagine-rich antigens, liver-stage antigens, octapeptide-repeat antigens and erythrocyte membrane-associated antigens—exhibit significantly elevated volatility compared with the other genes in the genome (Pw = 0.00013). In particular, the liver-stage antigens exhibit extremely significant elevated volatility (Table 1), in agreement with a comparative study that implicated liver-stage antigen for strong selection on the basis of its dN/dS ratio and a McDonald–Kreitman test15. These results confirm the ability of our method to detect those genes whose biology and sequence variation indicate strong diversifying selection.
The P. falciparum gene families named rifin, stevor and var are all thought to alter the parasite's antigenic surface proteins16. Although the roles of rifin and stevor are not understood, var-encoded proteins mediate adherence to host endothelial receptors, resulting in the sequestration of infected red cells17. Expression of var and cytoadherence are associated with disease severity and induction of protective antibodies3. The rifin or stevor families are not significantly over-represented among genes with high volatility. But the 71 var-encoded erythrocytic membrane proteins (EMP1) are significantly more volatile than other genes (Pw = 6 × 10-10). The elevated volatility of EMP1 genes reflects positive selection that is presumably driven by the known interactions between these proteins, which are expressed on the surface of infected red blood cells18, and the immune system of the host.
Despite the fact that EMP1 genes as a whole exhibit significantly elevated volatility, they constitute a heterogeneous family. A recent study has classified the EMP1 genes into biologically significant groupings: three major groups (called A, B and C) and two separate smaller groups (called A/B and B/C)19. None of the 30 most volatile EMP1 genes belong to the A or A/B groups. This segregation of genes is statistically significant (P < 5 × 10-6). We therefore propose that the A and A/B groups may experience less interaction with the human immune system than other EMP1 genes. Consistent with this hypothesis is the observation that recombinant CIDR domains from genes in group A do not bind to CD36, the major endothelial receptor for infected erythrocytes, whereas CIDR domains from groups B and C do bind CD36 (ref. 20).
Our estimates of the selective pressures across the P. falciparum genome will be useful for understanding host–pathogen interactions and for developing appropriately targeted vaccines21. The largest proportion of the highly volatile P. falciparum genes, however, are of unknown function (see Supplementary tables). This indicates the importance of further, targeted research on the biology of P. falciparum, particularly on genes under positive selection. We note that the P. falciparum genome also contains many genes, including some surface proteins, that exhibit strong signs (volatility P > 0.999) of purifying selection; some of these proteins might be attractive candidates for drug targets.
The identification of selective pressures across an entire genome also allows us to study the interplay between evolution and an organism's natural history, ecology or molecular processes. In Fig. 3 we compare volatility with gene expression data across the stages of P. falciparum's 48-hour intraerythrocytic development cycle4, and find a highly significant correlation between stage-specific gene expression and volatility: genes expressed in the ring stage show signs of positive selection relative to the rest of the genome, whereas genes expressed in the trophozoite and schizont stages show relatively more purifying selection (Fig. 3). These results shed light on the strength of immune pressure across the parasite's life cycle.

The lower panel shows the percentage of ring-stage (circles), trophozoite-stage (crosses) and schizont-stage (squares) parasites at each time point for which relative mRNA expression levels of 4,488 ORFs were measured4. The upper graph shows the Spearman correlation coefficient between gene expression level, Cy5/Cy3, and volatility P value among the genes of high (P < 0.001) and low (P > 0.999) volatility. Genes expressed during the ring stage are significantly correlated with high volatility, whereas trophozoite- and schizont-expressed genes generally exhibit low volatility. Of the 46 time points, 21 have significantly positive or negative correlations (P < 0.05 each).
When sufficient data are available, comparative sequence analysis is also a powerful method for estimating selective pressures. The most widely used measure, dN/dS, quantifies the ratio of non-synonymous to synonymous substitution rates6,22,23. Such techniques are usually applied to a small number of genes with orthologues in a large number of related species; power to detect selection is reduced when analysing fewer than six orthologous sequences24. At the genome-wide scale, comparative techniques are limited by the inability to find orthologues for all, or even most, genes in the genome being studied. (Fewer than half of the P. falciparum genes have identifiable orthologues for its closest fully sequenced relative, P. yoelii yoelii.)
We have seen that dN/dS and volatility detect elevated positive selection in the same gene families of M. tuberculosis2. For the purpose of a gene-by-gene comparison, we have analysed the genomes of three organisms related to M. tuberculosis strain CDC1551: M tuberculosis strain H37Rv, M. bovis and M. leprae. The set of M. tuberculosis genes with identifiable orthologues is significantly biased towards lower volatility (Pw = 10-6). Among the identifiable orthologues in pairwise genome comparisons, we find highly significant correlations between volatility P values and dN (P = 5 × 10-9; see Supplementary Methods). Therefore, even though analysis by codon volatility requires far fewer data than comparative techniques, it can produce a more complete, genome-wide account of selective pressures that is consistent with comparative techniques. Moreover, our method does not rely on some of the strongest assumptions inherent in comparative analyses (see Supplementary Methods).
Our method is not free of assumptions, however. In its simplest form, analysis by differential codon volatility rests on two underlying assumptions: (1) nucleotide mutations are unbiased and (2) the a priori probability of a codon's occurrence does not vary across the genome. The first assumption can easily be relaxed. When differential nucleotide mutation rates have been measured, such as a transition/transversion bias, these rates can be incorporated into the definition of codon volatility (see Methods).
The second assumption of our method holds approximately for most organisms, provided that the genome does not contain large variation in GC content. Although M. tuberculosis has different G and C contents on the leading and lagging replication strands25, our results remain unchanged if we analyse the genes on these two strands separately. The marked GC variation caused by isochores in mouse and human26 can be addressed by a separate analysis of volatility for those genes in each distinct region of GC content. Any other source of selection on synonymous codons that varies from gene to gene and that correlates with volatility will introduce some error into our estimates of relative selective pressures.
Genomic analysis by codon volatility has several important limitations. The foremost is that volatility P values are intrinsically relative. We cannot conclude that any gene is under positive selection in an absolute sense; rather, we can only conclude that some genes are under more positive, or less negative, selection than others. Moreover, codon volatility only detects selection on point mutations. Our method does not produce estimates of effective population sizes, divergence times, substitution rates or other evolutionary parameters that can be fitted through comparative analysis.
Finally, it is important to note that our method measures selective pressures in a fundamentally different way from comparative sequence analysis. Whereas comparative techniques consider sequence changes that have occurred since the divergence of the species being studied, volatility is based on the most recent substitution at each codon position in a single sequence. As a result, our method is preferentially sensitive to recent selective pressure, and it might even reflect selection on different timescales for different genes within the same genome; these timescales might be shorter or longer than those of comparative methods, depending on the species being compared and the orthologues that can be identified. Thus, the method of differential codon volatility complements comparative techniques. For a large range of fully sequenced organisms, a combination of volatility and comparative techniques will help to explain patterns of genome-wide evolution over a broad range of timescales.
Methods
Computing volatility P values
We define the volatility of codon c by the equation

where we sum over those non-stop codons ci that can mutate into c by a single point mutation. We use the simplest possible measure D: the Hamming metric, which equals zero if two amino acids are identical, and one otherwise. Equation (1) is similar to a measure recently used to analyse influenza virus genes27. Here, however, we disallow stop codons as potential ancestor codons.
Given a gene G, we define ν(G) as the summed volatility of the codons in the coding region of the gene G. To calculate the volatility P value of G, we compare the observed volatility ν(G) with a bootstrap distribution of 106 synonymous versions of the gene G. In each randomization trial, we construct a nucleotide sequence G′ that has the same translation as G but whose codons are drawn randomly according to the relative frequencies of synonymous codons in the genome as a whole. The P value for gene G is given by the proportion of the randomization trials in which ν(G′) exceeds or equals ν(G). Because there were no ties in our randomization trials, 1 - P is a P value that tests whether a gene is significantly less volatile than the genome as a whole. Software and a web implementation to perform this calculation are freely available (http://www.cgr.harvard.edu/volatility).
Our method of computing P values controls for both the length and amino-acid composition of each gene. If a gene contains many amino acids that can be encoded only by highly volatile codons (such as methionine), this feature will not bias the P value. The randomization procedure also controls for the nucleotide composition of the genome as well as any other source of genome-wide codon bias.
Equation (1) defines codon volatility under the assumption of equal mutation rates from each of the potential ancestor codons. When differential nucleotide mutation rates are known (for example, a transition/transversion bias), these rates can be incorporated into the definition of volatility by weighting the ancestor codons appropriately:

where ri is the rate of mutation from codon ci to codon c. Under a standard transition/transversion bias model, for example, ri will equal either κ or 1 depending upon whether codons ci and c differ by a transition or a transversion28. More detailed nucleotide mutation biases, including time-irreversible rates such as those measured for Drosophila29, can likewise be incorporated. The genomes of some mammals also exhibit strong dinucleotide mutational biases, particularly on CpG30. Such biases can also be incorporated into the definition of volatility by considering the flanking nucleotides of each codon when calculating the rates ri.
In the present study, we did not assume a transition/transversion bias because the strength of this bias is unknown for M. tuberculosis. Nevertheless, assuming that transitional mutations are twice as likely as transversional mutations does not significantly alter our results. (The volatility P values under κ = 1 versus κ = 2 are highly correlated; r = 0.94.) For P. falciparum, evidence suggests there is little or no bias towards transitions15.
Statistical methods
We have used the Wilcoxon test to compare the volatility P values within a group of genes (for example, the PPE genes) against the P values for the rest of genes in a genome. We denote the two-tailed significance of the Wilcoxon test by Pw. The PE, PPE and essential M. tuberculosis genes14 have median volatility P values of 0.06, 0.15 and 0.59, respectively. The EMP1 genes of P. falciparum have a median volatility P value of 0.17.
See also Supplementary Methods.
References
Zuckerkandl, E. & Pauling, L. Molecular disease, evolution and genic heterogeneity. in Horizons in Biochemistry (eds Kasha, M. & Pullman, B.) 189–225 (Academic, New York, 1962)
Fleischmann, R. D. et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J. Bacteriol. 184, 5479–5490 (2002)
Bull, P. et al. Parasite antigens on the infected red cell surface are targets for naturally acquired immunity to malaria. Nature Med. 4, 358–360 (1998)
Bozdech, Z. et al. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLOS Biol. 1, 1–16 (2003)
Gould, S. J. The Structure of Evolutionary Theory (Belknap, Cambridge, 2002)
Goldman, N. & Yang, Z. Codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–736 (1994)
Hudson, R. R., Kreitman, M. & Aguade, M. A test of neutral molecular evolution based on nucleotide data. Genetics 116, 153–159 (1987)
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989)
McDonald, J. H. & Kreitman, M. Adaptive protein evolution at the ADH locus in Drosophila. Nature 351, 652–654 (1991)
van Nimwegen, E., Crutchfield, J. & Huynen, M. Neutral evolution of mutational robustness. Proc. Natl Acad. Sci. USA 96, 9716–9820 (1999)
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998)
Ramakrishnan, L., Federspiel, N. A. & Falkow, S. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science 288, 1436–1439 (2000)
Skeiky, Y. A. W. et al. T cell expression cloning of a Mycobacterium tuberculosis gene encoding a protective antigen associated with the early control infection. J. Immunol. 165, 7140–7149 (2000)
Sassetti, C., Boyd, D. & Rubin, E. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 (2003)
Escalante, A. A., Lal, A. A. & Ayala, F. J. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics 149, 189–202 (1998)
Le Roch, K. G. et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301, 1503–1508 (2003)
Su, X. Z. et al. The large diverse gene family Var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum infected erythrocytes. Cell 82, 89–100 (1995)
Florens, L. et al. A proteomic view of the Plasmodium falciparum life cycle. Nature 419, 520–526 (2002)
Lavstsen, T., Salanti, A., Jensen, A., Arnot, D. & Theander, T. Sub-grouping of Plasmodium falciparum 3d7 var genes based on sequence analysis of coding and non-coding regions. Malaria J. 2, 1–14 (2003)
Robinson, B., Welch, T. & Smith, J. Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Mol. Microbiol. 47, 1265–1278 (2003)
Kissinger, J. C. et al. The Plasmodium genome database. Nature 419, 490–492 (2002)
Felsenstein, J. A likelihood approach to character weighting and what it tells us about parsimony and compatibility. Biol. J. Linn. Soc. 16, 183–196 (1981)
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Applic. Biosci. 13, 555–556 (1997)
Anisimova, M., Bielawski, J. P. & Yang, Z. The accuracy and power of likelihood ratio tests to detect positive selection at amino acid sites. Mol. Biol. Evol. 18, 1585–1592 (2001)
Lobry, J. R. & Seuoka, N. Asymmetric directional mutation pressures in bacteria. Genome Biol. 3, 0058.1–0058.14 (2002)
Bernardi, G. The human genome: Organization and evolutionary history. Annu. Rev. Genet. 29, 445–476 (1995)
Plotkin, J. B. & Dushoff, J. Codon bias and frequency-dependent selection on the hemagglutinin epitopes of influenza A virus. Proc. Natl Acad. Sci. USA 100, 7152–7157 (2003)
Yang, Z. & Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43 (2000)
Petrov, D. A. & Hartl, D. L. Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc. Natl Acad. Sci. USA 96, 1475–1479 (1999)
Zhang, Z. & Gerstein, M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 31, 5338–5348 (2003)
Acknowledgements
We thank A. Murray, S. Levin, D. Hartl, A. Clark, D. Petrov, C. Bustamante, L. Garwin, N. Friedman and P. Palese for their input during the preparation of this manuscript. We also thank the Harvard University Bauer Center for hosting the volatility computing server.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Supplementary information
Supplementary Table 1
Volatility p-values for the 4099 coding sequences of M. tuberculosis strain CDC1551. (XLS 369 kb)
Supplementary Table 2
Volatility p-values for the 5440 coding sequences of P. falciparum strain 3D7. (XLS 1002 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Plotkin, J., Dushoff, J. & Fraser, H. Detecting selection using a single genome sequence of M. tuberculosis and P. falciparum. Nature 428, 942–945 (2004). https://doi.org/10.1038/nature02458
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature02458
This article is cited by
-
Comparative genomics of Bacteria commonly identified in the built environment
BMC Genomics (2019)
-
Codon usage vis-a-vis start and stop codon context analysis of three dicot species
Journal of Genetics (2018)
-
Guanine and Cytosine at the Second Codon Position Influence Gene Expression in Cereals
Proceedings of the National Academy of Sciences, India Section B: Biological Sciences (2015)
-
Drift sometimes dominates selection, and vice versa: a reply to Clatterbuck, Sober and Lewontin
Biology & Philosophy (2014)
-
In Arabidopsis thaliana codon volatility scores reflect GC3 composition rather than selective pressure
BMC Research Notes (2012)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
