
Abstract
Anorexia nervosa (AN) is a complex neuropsychiatric disorder presenting with dangerously low body weight, and a deep and persistent fear of gaining weight. To date, only one genome-wide significant locus associated with AN has been identified. We performed an exome-chip based genome-wide association studies (GWAS) in 2158 cases from nine populations of European origin and 15 485 ancestrally matched controls. Unlike previous studies, this GWAS also probed association in low-frequency and rare variants. Sixteen independent variants were taken forward for in silico and de novo replication (11 common and 5 rare). No findings reached genome-wide significance. Two notable common variants were identified: rs10791286, an intronic variant in OPCML (P=9.89 × 10−6), and rs7700147, an intergenic variant (P=2.93 × 10−5). No low-frequency variant associations were identified at genome-wide significance, although the study was well-powered to detect low-frequency variants with large effect sizes, suggesting that there may be no AN loci in this genomic search space with large effect sizes.
Similar content being viewed by others


Introduction
Family studies of anorexia nervosa (AN) have consistently shown that first-degree relatives of AN sufferers have an increased risk of AN, compared with relatives of unaffected individuals.1, 2, 3, 4 Twin studies have estimated the heritability of AN at 56%,5 with the majority of remaining variance in liability attributed to non-shared environmental factors (38%).5
Three genome-wide association studies (GWAS) of AN have been conducted to date. The first comprised 1033 AN cases collected as part of the Price Foundation Genetic Study of Anorexia Nervosa and 3733 pediatric controls from the Children’s Hospital of Philadelphia.6 This study focused on common variation and identified 11 suggestive variants (P<1 × 10−5). None reached genome-wide significance in the primary analysis, although one variant (rs4479806) approached genome-wide significance in an associated secondary analysis. The second study (comprising 2907 cases and 14 860 controls) was carried out by the Genetic Consortium for AN, as part of the Wellcome Trust Case Control Consortium 3 (WTCCC3) effort.7 This study identified two suggestively associated variants (P<1 × 10−5). Notably, signals at P<1 × 10−5 were significantly more likely to have the same direction of effect in the replication as in the discovery cohorts (P=4 × 10−6), which implies that true signals exist within this data set, but that the study was underpowered for detection. Recently, a third study-meta-analyzed samples from both of these studies, as well as some novel cases, comprising a total of 3495 cases and 10 982 controls. To our knowledge, this study identified the first genome-wide significant locus for AN (index variant rs4622308, P=4.3 × 10−9).8
Both previous studies focused on common variation. Here, we conducted, to our knowledge, the first association study that also considered low frequency (minor allele frequency (MAF)<5%) and rare exonic variants in addition to common variation.
Materials and methods
Sample collections
We conducted a GWAS across nine discovery data sets (the majority overlapping with Genetic Consortium for AN, as part of the Wellcome Trust Case Control Consortium 3 (WTCCC3/WTCCC3 samples), resulting in a total of 2158 cases and 15 485 ancestrally matched controls (Table 1 and Figure 1). All AN cases were female. AN diagnosis was made via semistructured or structured interview, or population assessment strategy using Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria for AN. The amenorrhea criterion was not applied, as this has been shown not to be diagnostically relevant9 and has since been dropped from DSM-5.10, 11 All cases met criteria for lifetime AN.

Geographical distribution of samples across Europe. (a) Distribution of cases across Europe; 375 USA cases are not shown in this diagram. (b) Distribution of controls across Europe; 873 USA controls are not shown in this diagram.
Exclusion criteria included confounding medical diagnoses, for example, psychotic conditions, developmental delay or medical or neurological conditions causing weight loss.
Ancestry-matched controls were selected for each AN case set. Both male and female controls were used (Table 1). These were obtained either from existing collaborations, or through genotyping repository (dbGaP) access. Each site obtained ethical approval from the local ethics committee, and all participants provided written informed consent in accordance with the Declaration of Helsinki.
Population prevalence of AN in these populations ranged from 0.4 to 3% (refs 12, 13, 14, 15, 16, 17, 18; Table 1).
Genotyping
Cases were genotyped on either the ‘Infinium HumanCoreExome-12 BeadChip Kit (Illumina, San Diego, CA, USA),19 or the ‘Infinium HumanCoreExome-24 BeadChip Kit (Illumina),20 at the Wellcome Trust Sanger Institute. Where possible, controls were selected from existing studies with matching genotyping platforms to cases. Three control cohorts had been genotyped on the ‘Infinium HumanExome-12 BeadChip Kit’ (Table 1). To ameliorate potential confounding due to chip effects,21 chip-type quality control (QC) was carried out, and ~14 000 single-nucleotide polymorphisms (SNPs) removed.
Quality control
Genotypes were called using the GenCall22 and Zcall23 algorithms. At each of these genotype-calling stages, QC was performed for each population and for cases and controls separately (Supplementary Table 1). The final number of SNPs included in the analyses is given in Table 2.
Controlling for population stratification
In order to account for population stratification, a principal components analysis was carried out for each cohort separately using the smartpca software.24
Population outliers were identified by merging each population with central European 1000 Genomes data.25
Variance explained by each PC was plotted for each population. In order to be both conservative and consistent across populations, the first 10 principal components were included as covariates in the association testing.
Association testing
Unbalanced case–control ratios can lead to anticonservative P-value estimates.26 This study includes a number of unbalanced strata (Table 1). The likelihood ratio test has been shown to have low type-I error rate across both balanced and unbalanced cohorts,26 and was chosen as the association test for this study.
A lower cutoff of minor allele count of 5 and MAF of 0.1% was used. Association testing was performed for each cohort separately using SNPtest.27 In the cohorts with mixed sex controls (all except Italy and Norway), sex was also included as a covariate.
The standard genome-wide significance threshold of P⩽5 × 10−8 was applied.
Meta-analysis
Summary statistics across cohort were meta-analyzed using an inverse variance-based test in METAL.28 In order to test the heterogeneity of the results, Cochran’s Q and the I2 statistic were computed.
Assigning variants to genes
Variants identified associated at P⩽1 × 10−4 were assigned to genes using Ensembl (release 83; Ensembl Genome Browser).29, 30 For each variant, all predicted consequences (for example, missense, non-synonymous, and so on) and associated gene transcripts were downloaded and compared. Each variant was associated with only one predicted consequence and one Ensembl gene ID (Ensembl Genome Browser).29
Cluster plot checking
Cluster plots were created for all SNPs reaching P⩽1 × 10−4 in any analysis (cohort-specific or meta-analysis) using ScatterShot.31 SNPs were visually inspected for each cohort, and for cases and controls separately. In instances where multiple cohorts were merged (for example, UK cases), cluster plots were checked separately for each original cohort.
Burden testing
The potential aggregation of rare variants in cases compared with controls was investigated using a gene-based approach. Burden tests were carried out using the Zeggini–Morris burden test32 as implemented in rvtests (Rvtests - Genome Analysis Wiki).
All SNPs with MAF between 0.1 and 5% were included; similar to the single-point analysis, a lower bound of minor allele count=5 was used. A list of genes and locations was obtained from the UCSC genome browser (Table Browser: www.genome.ucsc.edu). All genes with at least two qualifying variants in at least two populations were used, resulting in a total of 9083 genes.
Burden tests were carried out for each population individually, and the results meta-analyzed using Stouffer’s method, weighted according to effective sample size.33
The genome-wide significance threshold for burden testing is computed in a similar manner to that for single-point analysis, using Bonferroni correction for the number of genes tested. This results in a genome-wide significance threshold of 5.5 × 10−6.
Pathway analysis
One of the key motivations of studying complex psychiatric disorders such as AN is the desire to unearth biological pathways underlying disease development. Pathway analysis was performed using summary statistics from the meta-analysis for the full data set.
Four pathway databases were used: the Kyoto Encyclopedia of Genes and Genomes (KEGG),34, 35 the Reactome pathway database (REACTOME),36 PANTHER pathway (PANTHER)37, 38 and the Gene Ontology database (GO).39, 40 These were curated to remove redundancy, resulting in a total set of 1836 pathways.
The analysis was run once on a merged set of 235 KEGG,34, 35 REACTOME36 and PANTHER37, 38 pathways, and once for the 1601 GO pathways.39, 40
Pathway analysis was carried out using MAGMA.41 MAGMA was selected for its ability to deal robustly with linkage disequilibrium (LD) between markers, correct for gene length and deal accurately with rare variants. To our knowledge, MAGMA was first used to annotate SNPs to genes. This analysis was repeated twice. In the first analysis, variants were assigned only to the gene they were in, resulting in 68.73% of the variants being assigned to 13 400 genes. In the second analysis, variants were assigned allowing a 20 kb window in both directions from the gene. This procedure included 75.44% of variants across 18 118 genes.
SNP P-values were used to create gene scores. The European panel of the 1000 Genomes project was used as a reference set to estimate LD between SNPs. The analysis also requires the sample size of the study to be specified; because of the unbalanced nature of the study, the effective sample sizes were given here.
Gene P-values were calculated using MAGMA.41 The top 10% of SNPs per gene were used. Significance was defined using a false discovery rate of 5%.42
There is a risk when assigning SNPs to genes using MAGMA that some highly associated SNP might be assigned to multiple overlapping genes, and thus distort pathway results. SNP–gene assignments were checked for all pathways that reached false discovery rate-corrected significance. No instances of SNPs being assigned to multiple genes were found across these pathways.
Replication
SNPs reaching P<1 × 10−4 in the discovery stage were prioritized for replication. In total, 16 SNPs were selected.
Replication was carried out using two data sets: one existing in silico data set and one set for de novo genotyping. The in silico data set came from an existing GWAS of AN,7 genotyped on the Illumina HumanHap610 platform. This data set included 1033 cases and 3733 controls. All cases included in this study were female. Controls were both male and female. The de novo replication cohort consisted of 266 self-volunteered female UK cases, collected through the charity Charlotte’s Helix (www.charlotteshelix.net). All participants were adults and had been diagnosed with AN by their clinician. In addition, all participants completed an online questionnaire based on the Structured Clinical Interview43 for the Diagnostic and Statistical Manual of Mental Disorders-IV Module H. The Structured Clinical Interview has been used extensively in epidemiological investigations. The Structured Clinical Interview eating disorder module was modified to capture information on lifetime history of eating disorders including AN, and includes questions on body mass index, age of onset, and experience of eating disorders. DNA from the saliva samples was extracted using standard protocols and was quantified using pico-green. Samples were genotyped on the Infinium HumanExome 12 Beadchip, genotypes were called using GenCall and Zcall algorithms and stringent QC was performed pre- and post-call. In all, 1500 ancestry-matched controls (55% female) were obtained from the UK Household Longitudinal Study.
De novo genotyping was performed using the iPLEX Assay and the MassARRAY System (Agena Bioscience, San Diego, CA, USA) (formerly Sequenom). Sample and SNP QC were carried out within each replication data set, using an 80% sample call rate and a 90% SNP call rate threshold, and a Hardy–Weinberg equilibrium threshold of 10−4. Five samples and one SNP were removed using these criteria.
Post-QC, 15 SNPs and 261 de novo cases remained. The de novo replication analysis therefore included 15 SNPs, 261 cases and 1500 controls. Genotypes for 12/16 SNPs were available in the in silico replication cohort, across 1033 in silico cases and 3733 controls.
Expression analysis
Gene expression data were obtained from the Genotype-Tissue Expression (GTex project) web portal, data release version 6 (dbGap Accession phs000424.v6.p1).44, 45, 46
Power
The sample sizes used in this study are small in the context of other psychiatric phenotypes. Power to identify genome-wide significant signals was calculated using Quanto.47, 48 This study is adequately powered to detect low-frequency alleles with large effect sizes and common alleles with substantial effect sizes (80% power to detect common alleles with odds ratio (OR)>1.5; low-frequency alleles with OR>2, Supplementary Figure 1).
Data availability
Genotypes of European cases included in this study are publicly available through the European Genome-Phenome Archive (EGA), under accession number EGAS00001000913, data set EGAD00010001043, with the exception of German and Dutch genotypes. Genotypes for cases from the United States of America may be obtained through dbGaP. Summary statistics are available for download from the PGC website (https://www.med.unc.edu/pgc/results-and-downloads).
Results
GWAS and replication meta-analyses
Association testing was performed separately for each of the nine discovery cohorts within this study (2158 cases, 15 485 controls), and the results were meta-analyzed. No inflation was seen in the QQ plot (Figure 2b). Six variants were identified with P<1 × 10−5, and nine additional variants with P<1 × 10−4 (Figure 2a and Supplementary Table 5). Of these, one variant approached genome-wide significance (exm860538/rs199965409, P=9.97 × 10−8), although this variant is polymorphic only in the Finnish population within these data sets, in the Exome Aggregation Consortium49 and in the 1000 Genomes project panel data.25 Variants with P<1 × 10−4 were taken forward for replication.

Results from discovery-phase meta-analyses. (a) Manhattan plot for meta-analyzed P-values, across all nine populations. (b) QQ plot (λ=0.94).
In total, 16 independent variants were selected for follow-up in one in silico cohort (1033 cases, 3733 controls) and one de novo genotyping cohort (261 cases, 15 000 controls). Of these, five were low frequency (MAF ~1%) and 11 were common frequency variants.
Twelve signals passed QC and were polymorphic in the de novo genotyping cohort, of which four were nominally significant (Supplementary Table 6; P<0.05, minimum P=0.001). Eight of twelve SNPs had the same direction of effect as in the discovery GWAS, including three of the four nominally significant variants.
Ten of the sixteen variants were present in the in silico cohort, of which six had the same direction of effect as in the discovery cohort, and one of these six was associated with P=0.02 (Supplementary Table 7).
On the basis of the number of SNPs taken forward for replication, we would not expect to see any variants reaching P<0.05 by chance. We also see a higher concordance in direction of effect between discovery and replication cohorts (7/10 in the in silico analysis, 8/12 in the de novo analysis) than might be expected by chance; however, the number of SNPs tested was too small to achieve statistical significance (P=0.17, P=0.19, one-sided binomial test).
Five SNPs had the same direction of effect across the meta-analyzed discovery cohort and both replication cohorts. No SNPs reached genome-wide significance in the final global meta-analysis. Two variants were associated with the same direction of effect across discovery and replication cohorts, and reached P<0.05 in at least one replication cohort (Table 3).
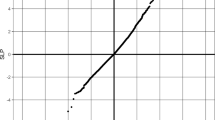
rs10791286 was associated with risk for AN across all discovery and replication cohorts (Figure 3a, global P=9.89 × 10−6, OR 0.84, 95% confidence interval 0.78–0.91). It resides in intron one of the opioid-binding protein/cell adhesion molecule-like (OPCML) gene. Data from the CommonMind Consortium project indicate that this variant is an eQTL for OPCML in the dorsolateral prefrontal cortex, and is associated with reduced expression (P=0.014 after correction for multiple testing).50 OPCML has a role in opioid-binding and opioid receptor function51, 52 and is expressed in a range of neuronal tissues, primarily the cerebellum and cerebellar hemispheres.44, 45, 46 OPCML has previous associations with body mass index,53 waist–hip ratio,54 visceral fat distribution55 and alcohol dependence,56 among other phenotypes. The variant itself has no previously reported associations in any phenotype.

Odds ratios for two notable single-nucleotide polymorphisms (SNPs) across discovery and replication cohorts. (a) rs10791286 and (b) rs7700147.
rs7700147 was associated with AN across all discovery and replication cohorts (global P=2.93 × 10−5, OR 1.2, 95% confidence interval: 1.1, 1.3; Figure 3b). It is an intergenic variant and has no previous associations.
Burden testing
Burden testing allows the contribution of multiple low-frequency variants to be aggregated across discrete units (for example, genes). Three genes were identified with P<1 × 10−4, although none reached genome-wide significance (Table 4). A further five genes reached P<1 × 10−4, but passed inclusion thresholds in one population only (Table 4), and as such are likely to be false-positives.
FAM96A has previously been associated with low-density lipoprotein levels and cholesterol57 and is primarily expressed in the liver, lymphocytes and adrenal gland.44, 45, 46 KIF7 has no previous phenotype associations and has generally low expression across a wide range of tissues.44, 45, 46 C6orf10 has previous associations with visceral fat55 and childhood obesity,58 as well as a number of autoimmune disorders.59, 60, 61, 62, 63, 64 C6orf10 is expressed in testes44, 45, 46 (see Discussion).
Biological pathways associated with AN
Allowing a 20 kb window for SNP to gene assignment identified two pathways significant at q<0.05: ‘Phospholipase activator’ and ‘GTP-rho binding’ (Table 5).
Using the strictest assignment method of SNPs to genes for the full data set, no pathways were significant after multiple-testing correction. The highest ranking pathway was ‘Calcium ion import’ (q-value=0.069).
Discussion
To our knowledge, this work constitutes the first examination of low frequency (<1% MAF) and rare exonic variation in AN in the context of a genome-wide scan. No low frequency or rare variant replicating associations were identified, although this study was well-powered to detect low-frequency variants with large effect sizes (Supplementary Figure 1). Although polymorphic only in the Finnish population, rs199965409 approached genome-wide significance. It is a non-synonymous variant with a MAF of 0.5% in the Finnish population.65, 66 The variant is within the WDR11 gene, which is associated with hypogonadotropic hypogonadism 14 with or without anosmia.67, 68, 69 The clinical features of the disease, such as delayed sexual maturation,68, 70, 71 suggest that it may be misdiagnosed or comorbid with AN, which may explain its association in the analysis.
Two notable, but common-frequency, signals were identified with consistent direction of effect across discovery and replication cohorts (rs10791286 and rs7700147). These variants had been removed from the first Genetic Consortium for AN, as part of the Wellcome Trust Case Control Consortium 3 (WTCCC3) AN GWAS because of poor cluster plots; therefore, we were not able to compare effect sizes between studies. Burden tests to investigate an aggregation of rare variants within genes rendered three potentially interesting genes, which require further replication.
Studying rare variation presents a range of challenges. The sample sizes required to identify rare variants with modest effect sizes are substantially larger than for common variants. Further, the MAF spectra seen across trans-European populations differ more for rare variants than for common variants, especially when considering genetically distant populations such as Finland and Italy.25 This can reduce the power to detect a signal and achieve replication. There are also many technical challenges to consider when conducting a rare variant study; for example, the inflation seen in association tests at low minor allele count26 and the increased error rate of calling algorithms when applied to rare variants22, 23, 72 We mitigated against the latter challenge by comprehensively examining cluster plots of >10 000 variants that surpassed a P-value threshold of P<1 × 10−4 in any analysis.
Of the genes potentially implicated through the single-point and burden test analyses, three have associations with metabolic and anthropometric phenotypes (OPCML, C6orf10 and FAM96a). OPCML has previously been associated with waist-to-hip ratio, while C6orf10 has associations with childhood obesity.58 FAM96A has been shown to be associated with metabolic phenotypes such as low-density lipoprotein and cholesterol levels. The associations of these three genes with metabolic and obesity-related phenotypes may indicate some roles for metabolic processes in AN development, although pathway analysis did not corroborate this observation. A growing body of evidence suggests involvement of metabolic processes in AN development, including appetite-satiety pathways, gut motility and gastric-emptying times.73, 74, 75, 76, 77, 78, 79 For example, application of the LD Score regression method revealed significant negative genetic correlations between AN and body mass index, insulin, glucose, and lipid phenotypes and significant positive genetic correlations between AN and HDL cholesterol phenotypes.1, 80
Notably, C6orf10 has been previously associated with childhood obesity.58 This finding is particularly interesting for a number of reasons. First, appetite and satiety dysregulation have been shown to be central to the development of childhood obesity.81, 82 In particular, reduced satiety responsiveness (experiencing an urge to eat despite internal ‘full’ signals) and heightened responsiveness to food have a role in increased adiposity. Aberrant responses to satiety signals and reduced responsiveness to food are also operative in AN, suggesting shared biological dysregulation between the two conditions.83, 84 Children with increased adiposity are at higher risk of eating disorders85 as they are more likely to engage in high-risk behaviors such as repeated and excessive dieting and erratic or overly rigid eating patterns.85, 86, 87, 88, 89 These children are also at higher risk of being bullied about their weight, which may increase weight and shape concerns, body dissatisfaction and a host of related risk factors for AN development.85, 86, 87, 88, 89, 90, 91, 92
The most significant pathway analysis association was with phospholipase activator pathways, which act to catalyze the hydrolysis of glycerophospholipids (GO:0016004 phospholipase activator activity). Phospholipase has a central role in the serotonin-triggered metabolism of arachidonic acid in the brain,93, 94, 95 which is a common target for antidepressants94, 95 such as lithium, carbamazepine (Tegretol), valproate and lamotrigine (Lamictal).96 These antidepressants have been shown to have varying efficacy in treating AN.97, 98, 99 Lithium has been used in treatment of AN (with varying success),97, 98, 99 while carbamazepine and valproate have been successfully used in individuals with complex comorbid eating disorder phenotypes.100, 101, 102, 103, 104 Finally, lamotrigine has been shown to significantly improve eating disorder and mood symptoms in individuals with binge-eating and purging behaviors.105
The second pathway identified as significantly associated with AN was GTP-rho binding. This pathway has a role in brain development, and is regulated by autism-susceptibility candidate gene 2 (AUTS2).106 This finding is consistent with the comorbidity between AN and autism.107 Moreover, individuals with AN may be socially withdrawn107 and exhibit elevated levels of autistic traits associated with lower social functioning.107, 108, 109 AUTS2 has also been well studied as a candidate gene for alcohol abuse,110 which is commonly comorbid with eating disorders.111 There is also a well-established link between GTP-rho activation and cognition.112 Mice with altered expression of genes regulating Rho-GTPases have been shown to have altered exploratory and anxiety-related behavior, decreased sociability and memory formation, and decreased body weight, among others.112 These findings are in line with some of the comorbidities and intermediate phenotypes noted in AN, for example, the high comorbidity with anxiety-related disorders.113
There is substantial evidence for the involvement of chromatin-modulating genes in the development of autism,114, 115, 116, 117, 118, 119 schizophrenia120, 121, 122, 123, 124 and body mass index changes.114 Given the comorbidity of these disorders with AN, and the potential overlap with autism indicated in the pathway analysis results, we tested for enrichment of chromatin-modulating genes in these results. We obtained a list of 340 genes involved in modifying chromatin accessibility and/or modifying histone marks from existing literature; of these, 30 reached nominal significance in our burden test, substantially more than expected by chance (binomial test, P=0.0026). Moreover, one of the variants identified in the global meta-analysis (exm540361) lays near a gene included in this list (UHRF1BP1). Together, these results may indicate a role for chromatin-modifying genes in AN, although more work will be needed to investigate this further.
A number of limitations should be borne in mind when evaluating these results. First, the sample size of this study is small. Psychiatric disorders in general require very large sample sizes in order to identify reliable genome-wide significant signals.125 The current study was powered to detect common variants with substantial OR, and rare variants conferring substantial increases in disease risk (OR>2). To our knowledge, this was the first time a study has specifically investigated the role of rare variation in AN, and the lack of low-frequency replicating findings may indicate that little advancements may be made in this particular genomic search space.
We did not see any overlap between the pathways identified here and those identified in the recent PGC pathway analysis;126 however, this may reflect the relatively small sample size of this study, as well as different pathway analysis methodologies used.
In this study we only examined female AN cases of European origin. It has been suggested that the genetics underlying AN development may be easier to assess in an all-male study,3 as there may be a greater genetic risk required to induce trait expression. The higher relative risk in male subjects may also reflect this.3 To date, this has not been possible because of the lower prevalence of the disorder in men, resulting in substantially smaller sample sizes. Moreover, if AN is heterogeneous between populations, in order to fully understand the genetic etiology of the disorder, it will be necessary to expand collection to include more diverse samples. Efforts are already underway in a number of Asian populations such as Taiwan, Japan, Korea and China, as well as some South American populations such as Argentina and Brazil.
A caveat to this study is that controls were not screened for AN, and that both male and female controls were used. Given the population prevalence of AN across population of European descent, ~80 female and ~10 male controls would be expected to have AN diagnoses. Given the low rate of treatment seeking in AN,127 it would not be possible to confidently screen population-based or previously existing control cohorts for AN.
The underlying biological etiology of AN is complex and has not been elucidated yet. Here we have identified a number of variants that warrant follow-up in larger sample sizes, and which point to a role for metabolic, appetite-related and obesity-related effects, in line with a growing body of evidence for metabolic involvement in AN development. Substantially increased sample sizes and detailed phenotyping to reduce heterogeneity will be necessary to empower the characterization of the genetic architecture of AN.
References
Strober M, Freeman R, Lampert C, Diamond J, Kaye W . Controlled family study of anorexia nervosa and bulimia nervosa: evidence of shared liability and transmission of partial syndromes. Am J Psychiatry 2000; 157: 393–401.
Strober M, Freeman R, Lampert C, Diamond J, Kaye W . Males with anorexia nervosa: a controlled study of eating disorders in first-degree relatives. Int J Eat Disord 2001; 29: 263–269.
Lilenfeld LR, Kaye WH, Greeno CG, Merikangas KR, Plotnicov K, Pollice C et al. A controlled family study of anorexia nervosa and bulimia nervosa: psychiatric disorders in first-degree relatives and effects of proband comorbidity. Arch Gen Psychiatry 1998; 55: 603–610.
Bulik CM, Slof-Op’t Landt MCT, van Furth EF, Sullivan PF . The genetics of anorexia nervosa. Annu Rev Nutr 2007; 27: 263–275.
Bulik CM, Sullivan PF, Tozzi F, Furberg H, Lichtenstein P, Pedersen NL . Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry 2006; 63: 305–312.
Wang K, Zhang H, Bloss CS, Duvvuri V, Kaye W, Schork NJ et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry 2011; 16: 949–959.
Boraska V, Franklin CS, Floyd JAB, Thornton LM, Huckins LM, Southam L et al. A genome-wide association study of anorexia nervosa. Mol Psychiatry 2014; 19: 1085–1094.
Duncan L, Yilmaz Z, Gaspar H, Walters R, Goldstein J, Anttila V et al. Significant locus and metabolic genetic correlation revealed in genome-wide association study of anorexia nervosa. Am J Psychiatry 2017 (in press).
Poyastro Pinheiro A, Thornton LM, Plotonicov KH, Tozzi F, Klump KL, Berrettini WH et al. Patterns of menstrual disturbance in eating disorders. Int J Eat Disord 2007; 40: 424–434.
American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders. 5th edn. American Psychiatric Association: Arlington, 2013.
American Psychiatric Association in Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association, 2013; doi:10.1176/appi.books.97808.
Preti A, de Girolamo G, Vilagut G, Alonso J, de Graaf R, Bruffaerts R et al. The epidemiology of eating disorders in six European countries: results of the ESEMeD-WMH project. J Psychiatr Res 2009; 43: 1125–1132.
Keski-Rahkonen A, Hoek HW, Susser ES, Linna MS, Sihvola E, Raevuori A et al. Epidemiology and course of anorexia nervosa in the community. Am J Psychiatry 2007; 164: 1259–1265.
Rathner G, Messner K . Detection of eating disorders in a small rural town: an epidemiological study. Psychol Med 1993; 23: 175–184.
Bijl RV, Ravelli A, van Zessen G . Prevalence of psychiatric disorder in the general population: results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Soc Psychiatry Psychiatr Epidemiol 1998; 33: 587–595.
Kringlen E, Torgersen S, Cramer V . A Norwegian Psychiatric Epidemiological Study. Am J Psychiatry 2001; 158: 1091–1098.
PriceWaterhouseCoopers & BEAT. The costs of eating disorders Social, health and economic impacts. 2015; https://www.b-eat.co.uk/assets/000/000/302/The_costs_of_eating_disorders_Final_original.pdf?1424694814.
Hudson JI, Hiripi E, Pope HG, Kessler RC . The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry 2007; 61: 348–358.
Illumina. Infinium ® Human Exome-12 v1.2 BeadChip. Data Sheet: Genotyping 2015. Available at: http://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet%7B_%7Dhumanexome%7B_%7Dbeadchips.pdf.
Illumina. Infinium ® CoreExome-24 v1.1 BeadChip. Data Sheet: Genotyping 2015; pp 1-2. Available at: http://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet%7B_%7Dhuman%7B_%7Dcore%7B_%7Dexome%7B_%7Dbeadchip.pdf.
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2014; 517: 327–332.
Ritchie ME, Liu R, Carvalho BS, Irizarry RA . Comparing genotyping algorithms for Illumina’s Infinium whole-genome SNP BeadChips. BMC Bioinformatics 2011; 12: 68.
Goldstein JI, Crenshaw A, Carey J, Grant GB, Maguire J, Fromer M et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics 2012; 28: 2543–2545.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Ma C, Blackwell T, Boehnke M, Scott LJ . Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet Epidemiol 2013; 37: 539–550.
Marchini J, Howie B . Genotype imputation for genome-wide association studies. Nat Rev Genet 2010; 11: 499–511.
Willer CJ, Li Y, Abecasis GR . METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–2191.
Rios D, McLaren WM, Chen Y, Birney E, Stabenau A, Flicek P et al. A database and API for variation, dense genotyping and resequencing data. BMC Bioinformatics 2010; 11: 238.
Bronwen LA, Sarah A, Daniel B, Laura C, Valery C, Susan F et al. The Ensembl gene annotation system. Database (Oxford) 2016; 2016: baw093; doi: 10.1093/database/baw093.
Rayner NW, Robertson N, McCarthy MI. ScatterShot; a Java program for creating cluster plots from Affymetrix and Illumina genotype data. American Society of Human Genetics Meeting (Poster Presentation), American Society of Human Genetics Meeting, Boston Convention & Exhibition Center (BCEC); 22–26 October 2013..
Morris AP, Zeggini E . An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 2010; 34: 188–193.
Stouffer SA . The American Soldier (Vol. 1): Adjustment During Army Life. Princeton University Press: Princeton, NJ, 1949.
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M . Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 2014; 42 (Database issue): D199–D205.
Kanehisa M, Goto S . KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28: 27–30.
Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G et al. The reactome pathway knowledgebase. Nucleic Acids Res 2014; 42 (Database issue): D472–D477.
Mi H, Muruganujan A, Thomas PD . PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 2013; 41 (Database issue): D377–D386.
Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 2003; 13: 2129–21241.
The Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res 2014; 43 (D1): D1049–D1056.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet Nat 2000; 25: 25–29.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D . MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 2015; 11: e1004219.
Benjamin Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 1995; 57: 289–300.
First MB, Spitzer RL, Gibbon M, Williams JBW . Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Non-patient Edition (SCID-I/NP) Biometrics Research. New York State Psychiatric Institute: New York, NY, USA, 2002.
Ardlie KG, Deluca DS, Segre AV, Sullivan TJ, Young TR, Gelfand ET et al. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–660.
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature Genetics 2013; 45: 580–585.
Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M et al. The human transcriptome across tissues and individuals. Science 2015; 348: 660–665.
Gauderman WJ . Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med 2002; 21: 35–50.
Gauderman WJ, Morrison JM . QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies, 2006. Available at: http://biostats.usc.edu/software.
Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv (Cold Spring Harbor Labs Journals, 2015); doi:10.1101/030338.
Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 2016; 19: 1442–1453.
OPCML Symbol Report | HUGO Gene Nomenclature Committee. Available at: http://www.genenames.org/cgi-bin/gene_symbol_report?hgnc_id=8143.
OPCML opioid binding protein/cell adhesion molecule-like [Homo sapiens (human)] - Gene - NCBI. Available at: https://www.ncbi.nlm.nih.gov/gene/4978.
Fox CS, Heard-Costa N, Cupples LA, Dupuis J, Vasan RS, Atwood LD . Genome-wide association to body mass index and waist circumference: the Framingham Heart Study 100K project. BMC Med Genet 2007; 8 (Suppl 1): S18.
Liu C-T, Monda KL, Taylor KC, Lange L, Demerath EW, Palmas W et al. Genome-wide association of body fat distribution in African ancestry populations suggests new loci. PLoS Genet 2013; 9: e1003681.
Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet 2012; 8: e1002695.
McGue M, Zhang Y, Miller MB, Basu S, Vrieze S, Hicks B et al. A genome-wide association study of behavioral disinhibition. Behav Genet 2013; 43: 363–373.
Kathiresan S, Manning AK, Demissie S, D’Agostino RB, Surti A, Guiducci C et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet 2007; 8 (Suppl 1): S17.
Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS ONE 2012; 7: e51954.
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703–707.
Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med 2009; 360: 2544–2555.
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 2008; 358: 900–909.
Urabe Y, Ochi H, Kato N, Kumar V, Takahashi A, Muroyama R et al. A genome-wide association study of HCV-induced liver cirrhosis in the Japanese population identifies novel susceptibility loci at the MHC region. J Hepatol 2013; 58: 875–882.
Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357: 851–862.
Negi S, Juyal G, Senapati S, Prasad P, Gupta A, Singh S et al. A genome-wide association study reveals ARL15, a novel non-HLA susceptibility gene for rheumatoid arthritis in North Indians. Arthritis Rheum 2013; 65: 3026–3035.
SISu. SISu Project. Available at http://www.sisuproject.fi/. Accessed on 13 May 2016.
Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet 2014; 10: e1004494.
Izumi Y, Suzuki E, Kanzaki S, Yatsuga S, Kinjo S, Igarashi M et al. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril 2014; 102: 1130–1136, e3.
Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V, Beckers A . Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol 2014; 5: 109.
Mao J, Chen R, Wu X, Liu Z, Zheng J, Wang X et al. Association of the extra-gonadal manifestations with different pathogenic gene mutations in male patients with congenital hypogonadotropic hypogonadism. Zhonghua Yi Xue Za Zhi 2015; 95: 3424–3427.
Kim SH . Congenital hypogonadotropic hypogonadism and kallmann syndrome: past, present, and future. Endocrinol Metab 2015; 30: 456–466.
Fenichel P . Delayed puberty. Endocr Dev 2012; 22: 138–159.
Shah TS, Liu JZ, Floyd JAB, Morris JA, Wirth N, Barrett JC et al. optiCall: a robust genotype-calling algorithm for rare, low-frequency and common variants. Bioinformatics 2012; 28: 1598–1603.
McCallum RW, Grill BB, Lange R, Planky M, Glass EE, Greenfeld DG . Definition of a gastric emptying abnormality in patients with anorexia nervosa. Dig Dis Sci 1985; 30: 713–722.
Szmukler GI, Young GP, Lichtenstein M, Andrews JT . A serial study of gastric emptying in anorexia nervosa and bulimia. Aust N Z J Med 1990; 20: 220–225.
Benini L, Todesco T, Dalle Grave R, Deiorio F, Salandini L, Vantini I . Gastric emptying in patients with restricting and binge/purging subtypes of anorexia nervosa. Am J Gastroenterol 2004; 99: 1448–1454.
Humphries L, Shih WJ . Gastric emptying time in anorexia nervosa and bulimia. Arch Surg 1988; 123: 783.
Kamal N, Chami T, Andersen A, Rosell FA, Schuster MM, Whitehead WE . Delayed gastrointestinal transit times in anorexia nervosa and bulimia nervosa. Gastroenterology 1991; 101: 1320–1324.
Stacher G, Kiss A, Wiesnagrotzki S, Bergmann H, Höbart J, Schneider C . Oesophageal and gastric motility disorders in patients categorised as having primary anorexia nervosa. Gut 1986; 27: 1120–1126.
García Aroca J, Alonso Calderón JL, García Redondo C, Rollán Villamarín V . Anorexia nervosa or somatic disease. Cir Pediatr 2001; 14: 98–102.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR et al, Consortium R. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015; 47: 1236–1241.
Llewellyn CH, Trzaskowski M, van Jaarsveld CHM, Plomin R, Wardle J . Satiety mechanisms in genetic risk of obesity. JAMA Pediatr 2014; 168: 338–344.
van Jaarsveld CHM, Boniface D, Llewellyn CH, Wardle J . Appetite and growth: a longitudinal sibling analysis. JAMA Pediatr 2014; 168: 345–350.
Avena NM, Bocarsly ME . Dysregulation of brain reward systems in eating disorders: neurochemical information from animal models of binge eating, bulimia nervosa, and anorexia nervosa. Neuropharmacology 2012; 63: 87–96.
Kaye WH, Wierenga CE, Bailer UF, Simmons AN, Bischoff-Grethe A . Nothing tastes as good as skinny feels: the neurobiology of anorexia nervosa. Trends Neurosci 2013; 36: 110–120.
Cattarin JA, Thompson JK . A three-year longitudinal study of body image, eating disturbance, and general psychological functioning in adolescent females. Eat Disord 1994; 2: 114–125.
Fairburn CG, Cooper Z, Doll HA, Davies BA . Identifying dieters who will develop an eating disorder: a prospective, population-based study. Am J Psychiatry 2005; 162: 2249–2255.
Patton GC, Selzer R, Coffey C, Carlin JB, Wolfe R . Onset of adolescent eating disorders: population based cohort study over 3 years. Br Med J 1999; 318: 765–768.
Santonastaso P, Friederici S, Favaro A . Full and partial syndromes in eating disorders: a 1-year prospective study of risk factors among female students. Psychopathology 32: 50–56.
Rohde P, Stice E, Marti CN . Development and predictive effects of eating disorder risk factors during adolescence: Implications for prevention efforts. Int J Eat Disord 2015; 48: 187–198.
Gardner RM, Stark K, Friedman BN, Jackson NA . Predictors of eating disorder scores in children ages 6 through 14: a longitudinal study. J Psychosom Res 2000; 49: 199–205.
Killen JD, Taylor CB, Hayward C, Haydel KF, Wilson DM, Hammer L et al. Weight concerns influence the development of eating disorders: a 4-year prospective study. J Consult Clin Psychol 1996; 64: 936–940.
Copeland WE, Bulik CM, Zucker N, Wolke D, Lereya ST, Costello EJ . Does childhood bullying predict eating disorder symptoms? A prospective, longitudinal analysis. Int J Eat Disord 2015; 48: 1141–1149.
Felder CC, Kanterman RY, Ma AL, Axelrod J . Serotonin stimulates phospholipase A2 and the release of arachidonic acid in hippocampal neurons by a type 2 serotonin receptor that is independent of inositolphospholipid hydrolysis. Proc Natl Acad Sci USA 1990; 87: 2187–2191.
Lee H-J, Rao JS, Chang L, Rapoport SI, Kim H-W . Chronic imipramine but not bupropion increases arachidonic acid signaling in rat brain: is this related to ‘switching’ in bipolar disorder? Mol Psychiatry 2010; 15: 602–614.
Bazinet RP . Is the brain arachidonic acid cascade a common target of drugs used to manage bipolar disorder? Biochem Soc Trans 2009; 37 (Pt 5): 1104–1109.
Rao JS, Rapoport SI . Mood-stabilizers target the brain arachidonic acid cascade. Curr Mol Pharmacol 2009; 2: 207–214.
Brewerton TD . Antipsychotic agents in the treatment of anorexia nervosa: neuropsychopharmacologic rationale and evidence from controlled trials. Curr Psychiatry Rep 2012; 14: 398–405.
Suárez-Pinilla P, Peña-Pérez C, Arbaizar-Barrenechea B, Crespo-Facorro B, Del Barrio JAG, Treasure J et al. Inpatient treatment for anorexia nervosa: a systematic review of randomized controlled trials. J Psychiatr Pract 2015; 21: 49–59.
Milano W, De Rosa M, Milano L, Riccio A, Sanseverino B, Capasso A . The pharmacological options in the treatment of eating disorders. ISRN Pharmacol 2013; 2013: 352865.
McElroy SL, Kotwal R, Keck PE, Akiskal HS . Comorbidity of bipolar and eating disorders: distinct or related disorders with shared dysregulations? J Affect Disord 2005; 86: 107–127.
McElroy SL, Guerdjikova AI, Martens B, Keck PE, Pope HG, Hudson JI . Role of antiepileptic drugs in the management of eating disorders. CNS Drugs 2009; 23: 139–156.
Cordás TA, Tavares H, Calderoni DM, Stump GV, Ribeiro RB . Oxcarbazepine for self-mutilating bulimic patients. Int J Neuropsychopharmacol 2006; 9: 769–771.
Tachibana N, Sugita Y, Teshima Y, Hishikawa Y . A case of anorexia nervosa associated with epileptic seizures showing favorable responses to sodium valproate and clonazepam. Jpn J Psychiatry Neurol 1989; 43: 77–84.
Tor PC, Lee EL . Treatment emergent mania responding to valproate in a Chinese female adolescent population with eating disorders: a case series. Eur Eat Disord Rev 2008; 16: 421–426.
Trunko ME, Schwartz TA, Marzola E, Klein AS, Kaye WH . Lamotrigine use in patients with binge eating and purging, significant affect dysregulation, and poor impulse control. Int J Eat Disord 2014; 47: 329–334.
Hori K, Nagai T, Shan W, Sakamoto A, Taya S, Hashimoto R et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep 2014; 9: 2166–2179.
Zucker NL, Losh M, Bulik CM, LaBar KS, Piven J, Pelphrey KA . Anorexia nervosa and autism spectrum disorders: guided investigation of social cognitive endophenotypes. Psychol Bull 2007; 133: 976–1006.
Baron-Cohen S, Jaffa T, Davies S, Auyeung B, Allison C, Wheelwright S . Do girls with anorexia nervosa have elevated autistic traits? Mol Autism 2013; 4: 24.
Koch SV, Larsen JT, Mouridsen SE, Bentz M, Petersen L, Bulik C et al. Autism spectrum disorder in individuals with anorexia nervosa and in their first- and second-degree relatives: Danish nationwide register-based cohort-study. Br J Psychiatry 2015; 206: 401–407.
Schumann G, Coin LJ, Lourdusamy A, Charoen P, Berger KH, Stacey D et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci USA 2011; 108: 7119–7124.
Bulik CM, Klump KL, Thornton L, Kaplan AS, Devlin B, Fichter MM et al. Alcohol use disorder comorbidity in eating disorders: a multicenter study. J Clin Psychiatry 2004; 65: 1000–1006.
De Filippis B, Romano E, Laviola G . Aberrant Rho GTPases signaling and cognitive dysfunction: in vivo evidence for a compelling molecular relationship. Neurosci Biobehav Rev 2014; 46 (Pt 2): 285–301.
Kaye WH, Bulik CM, Thornton L, Barbarich N, Masters K . Comorbidity of anxiety disorders with anorexia and bulimia nervosa. Am J Psychiatry 2004; 161: 2215–2221.
Loviglio MN, Leleu M, Männik K, Passeggeri M, Giannuzzi G, van der Werf I et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol Psychiatry 2016.
Ciernia AV, LaSalle J . The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat Rev Neurosci 2016; 17: 411–423.
Barnard RA, Pomaville MB, O’Roak BJ . Mutations and modeling of the chromatin remodeler CHD8 define an emerging autism etiology. Front Neurosci 2015; 9: 477.
Shang L, Henderson LB, Cho MT, Petrey DS, Fong C-T, Haude KM et al. De novo missense variants in PPP2R5D are associated with intellectual disability, macrocephaly, hypotonia, and autism. Neurogenetics 2016; 17: 43–49.
Grayson DR, Guidotti A . Merging data from genetic and epigenetic approaches to better understand autistic spectrum disorder. Epigenomics 2016; 8: 85–104.
Sanders SJ . First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev 2015; 33: 80–92.
Eckart N, Song Q, Yang R, Wang R, Zhu H, McCallion AS et al. Functional characterization of schizophrenia-associated variation in CACNA1C. PLoS ONE 2016; 11: e0157086.
Fryland T, Christensen JH, Pallesen J, Mattheisen M, Palmfeldt J, Bak M et al. Identification of the BRD1 interaction network and its impact on mental disorder risk. Genome Med 2016; 8: 53.
González-Peñas J, Amigo J, Santomé L, Sobrino B, Brenlla J, Agra S et al. Targeted resequencing of regulatory regions at schizophrenia risk loci: role of rare functional variants at chromatin repressive states. Schizophr Res 2016; 174: 10–16.
Maschietto M, Tahira AC, Puga R, Lima L, Mariani D, Paulsen B et al. Co-expression network of neural-differentiation genes shows specific pattern in schizophrenia. BMC Med Genomics 2015; 8: 23.
Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 2014; 84: 997–1008.
McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9: 356–369.
Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015; 18: 199–209.
Keski-Rahkonen A, Mustelin L . Epidemiology of eating disorders in Europe. Curr Opin Psychiatry 2016; 29: 340–345.
Acknowledgements
Klarman Family Foundation: This study was supported by a grant from the Klarman Family Foundation.
Wellcome Trust Case Control Consortium: This work was funded by a grant from the WTCCC3 WT088827/Z/09 entitled ‘A genomewide association study of anorexia nervosa’.
Acknowledgements for specific authors: Eleftheria Zeggini is supported by the Wellcome Trust (098051). Lorraine Southam is supported by the Wellcome Trust (098051). Laura Huckins acknowledges Wellcome Trust (098051) and the MRC (MR/J500355/1). Cynthia Bulik acknowledges funding from the Swedish Research Council (VR Dnr: 538–2013–8864). Esther Walton is supported by the German Research Foundation (Wa 3635/1–1). Zeynep Yilmaz is funded by NIH K01MH109782. Allan Kaplan, Robert Levitan and James Kennedy acknowledge support from Ontario Mental Health Foundation for the collection of samples. Jaakko Kaprio is suppoted by the Academy of Finland (263278).
Charlotte’s Helix: We thank all the probands and parents from the parent-led group, Charlotte’s Helix. This charity was set up by Charlotte Bevan, after her daughter was diagnosed with anorexia nervosa. The charity is deeply committed to supporting biological work, particularly genetics, to help understand anorexia and has set up a database of patients’ names and details. Charlotte’s Helix has been collaborating closely with the King’s College London (KCL) team since 2013 by providing the database of probands, some funding and publicizing the scientific projects through regular blogs on its website and social media.
We gratefully acknowledge the participation of NIHR BRC South London and the Maudsley NHS Foundation Trust (SLaM) BioResource volunteers, and thank the NIHR BRC SLaM BioResource centre and staff for their contribution.
This paper represents independent research partly funded by the National Institute for Health Research (NIHR) Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.' We gratefully acknowledge capital equipment funding from the Maudsley Charity (Grant Ref. 980) and Guy’s and St Thomas’s Charity (Grant Ref. STR130505).
dbGap: We obtained High Density SNP Association Analysis of Melanoma: Case–Control and Outcomes Investigation data set through dbGaP (dbGaP Study Accession: phs000187.v1.p1). Research support to collect data and develop an application to support this project was provided by 3P50CA093459, 5P50CA097007, 5R01ES011740 and 5R01CA133996.
TEENAGE (TEENs of Attica: Genes and Environment): This work was funded by the Wellcome Trust (098051) and has been co-financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program ‘Education and Lifelong Learning’ of the National Strategic Reference Framework (NSRF)— Research Funding Program: Heracleitus II. Investing in knowledge society through the European Social Fund. We thank all study participants and their families as well as all volunteers for their contribution in this study. We thank the following staff from the Sample Management and Genotyping Facilities at the Wellcome Trust Sanger Institute for sample preparation, QC and genotyping: Dave Jones, Doug Simpkin, Emma Gray, Hannah Blackburn and Sarah Edkins.
UKHLS (The UK Household Longitudinal Study): The UK Household Longitudinal Study (UKHLS) is led by the Institute for Social and Economic Research at the University of Essex and is funded by the Economic and Social Research Council. The survey was conducted by NatCen, and the genome-wide scan data were analyzed and deposited by the Wellcome Trust Sanger Institute. Information on how to access the data can be found on the Understanding Society website https://www.understandingsociety.ac.uk/.
Exome Aggregation Consortium: We thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
CommonMind Consortium: CommonMind Consortium Data were used in this manuscript. These data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F Hoffman-La Roche and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138 and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner and Barbara Lipska (NIMH)’.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
GB has received grant funding and consultancy fees from Eli Lilly. DD is speaker, consultant or on advisory boards of various pharmaceutical companies, including AstraZeneca, Boehringer, Bristol Myers Squibb, Eli Lilly, 28 Genesis Pharma, GlaxoSmithKline, Janssen, Lundbeck, Organon, Sanofi, UniPharma and Wyeth, and he has unrestricted grants from Lilly and AstraZeneca as director of the Sleep Research Unit of Eginition Hospital (National and Kapodistrian University of Athens, Greece). AK is on the Shire Canada BED Advisory Board. JK is a member of SAB of AssurexHealth Inc (unpaid). ML has received lecture honoraria from Lundbeck, AstraZeneca and Biophausia Sweden, and served as scientific consultant for EPID Research Oy. There exists no other equity ownership, profit-sharing agreements, royalties, or patents. PS is scientific advisor to Pfizer, Inc. JT received an honorarium for speaking at a diabetic conference for Lilly and royalties from a published book. The remaining authors declare no conflicts of interest.
Additional information
RAH Adan37,38, L Alfredsson39, T Ando40, OA Andreassen41,42, H Aschauer43, JH Baker44, JC Barrett45, V Bencko46, AW Bergen47, WH Berrettini48, A Birgegård49,50, C Boni51, V Boraska Perica45,52, H Brandt53, G Breen54,55, CM Bulik56,57, L Carlberg58, M Cassina59, S Cichon60,61,62, M Clementi59, S Cohen-Woods63,64, J Coleman65, RD Cone66, P Courtet67, S Crawford53, S Crow68, J Crowley57,69, UN Danner38, OSP Davis70, M de Zwaan71, G Dedoussis72, D Degortes73, JE DeSocio74, DM Dick75, D Dikeos76, C Dina77, B Ding78, M Dmitrzak-Weglarz79, E Docampo80,81, L Duncan82,83, K Egberts84, S Ehrlich85,86, G Escaramís87, T Esko88,89,90, T Espeseth91, X Estivill92, A Favaro73, F Fernández-Aranda93,94, MM Fichter95, C Finan96, K Fischer97, JAB Floyd45, L Foretova98, M Forzan99, CS Franklin45, S Gallinger100,101, G Gambaro102, HA Gaspar54, I Giegling103, F Gonidakis104, P Gorwood105, M Gratacos87, S Guillaume67, Y Guo106, H Hakonarson107,108, KA Halmi109, K Hatzikotoulas45, J Hauser110, J Hebebrand111, S Helder112,113, S Herms114, B Herpertz-Dahlmann115, W Herzog116, CE Hilliard117, A Hinney111, C Hübel118, LM Huckins45,119, JI Hudson120, J Huemer121, H Inoko121, V Janout122, S Jiménez-Murcia123,124, C Johnson125, A Julià126, A Juréus127, G Kalsi128, D Kaminska129, AS Kaplan130, J Kaprio131,132, L Karhunen133, A Karwautz119, MJH Kas134,135, W Kaye136, JL Kennedy137, A Keski-Rahkonen138, K Kiezebrink139, L Klareskog140, KL Klump141, GPS Knudsen142, BPC Koeleman143, D Koubek119, MC La Via44, M Landén127,144, S Le Hellard145,146, RD Levitan147, D Li127, P Lichtenstein127, L Lilenfeld148, J Lissowska149, A Lundervold150,151, P Magistretti152,153, M Maj154, K Mannik155,156, S Marsal126, N Martin157, M Mattingsdal158,159, S McDevitt160, P McGuffin161, E Merl121, A Metspalu88, I Meulenbelt162, N Micali163, J Mitchell164, K Mitchell165,166, P Monteleone167,168, AM Monteleone154, P Mortensen169, MA Munn-Chernoff44, M Navratilova98, I Nilsson170,171, C Norring49,50, I Ntalla172, RA Ophoff37,173, JK O'Toole174, A Palotie83,175, J Pantel66,176, H Papezova177, D Pinto178, R Rabionet92, A Raevuori179,180, A Rajewski181, N Ramoz51, NW Rayner45,182,183, T Reichborn-Kjennerud184, S Ripatti185, M Roberts186, A Rotondo187, D Rujescu103,188, F Rybakowski189, P Santonastaso73, A Scherag190, SW Scherer191,192, U Schmidt186, NJ Schork193, A Schosser194, L Slachtova195, R Sladek196, PE Slagboom197, MCT Slof-Op 't Landt198, A Slopien199, N Soranzo200,201,202, L Southam45, VM Steen203,204, E Strengman205, M Strober206,207, PF Sullivan57,208, JP Szatkiewicz69, N Szeszenia-Dabrowska209, I Tachmazidou45, E Tenconi210, LM Thornton44, A Tortorella154,211, F Tozzi212, J Treasure186, A Tsitsika213, K Tziouvas214, AA van Elburg134,215, EF van Furth216,217, G Wagner121, E Walton85,86,218,219, H Watson220,221, H-E Wichmann222, E Widen175, DB Woodside223,224, J Yanovski225, S Yao127, Z Yilmaz44, E Zeggini45, S Zerwas44 and S Zipfel226 37University Medical Center Utrecht, Utrecht, The Netherlands; 38Altrecht Eating Disorders Rintveld, Zeist, The Netherlands; 39Institute of Environmental Medicine, Karolinska Institute, Stockholm, Sweden; 40Department of Psychosomatic Research, National Institute of Mental Health, National Center of Neurology and Psychiatry, Tokyo, Japan; 41Division of Mental Health and Addiction, NORMENT, K.G. Jebsen Centre for Psychosis Research, Oslo University Hospital, Oslo, Norway; 42Institute of Clinical Medicine, University of Oslo, Oslo, Norway; 43Biopsychosocial Corporation, Vienna, Austria; 44Department of Psychiatry, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 45Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, UK; 46Institute of Hygiene and Epidemiology, First Faculty of Medicine, Charles University, Prague, Czech Republic; 47Biorealm Research, Culver City, CA, USA; 48Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA; 49Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden; 50Stockholm Health Care Services, Stockholm County Council, Stockholm, Sweden; 51INSERM U984, Centre of Psychiatry and Neuroscience, Paris, France; 52University of Split School of Medicine, Split, Croatia; 53The Center for Eating Disorders at Sheppard Pratt, Baltimore, MD, USA; 54Social Genetic and Developmental Psychiatry, King's College London, London, UK; 55NIHR BRC for Mental Health, Institute of Psychiatry, Psychology and Neuroscience, King's College London, SLaM NHS Trust, London, UK; 56Department of Psychiatry and Nutrition, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 57Karolinska Institutet, Stockholm, Sweden; 58Department of Psychiatry, Medical University of Vienna, Vienna, Austria; 59Clinical Genetics Unit, Department of Women’s and Children’s Health, University of Padova, Padova, Italy; 60Department of Biomedicine, University Hospital Basel, Basel, Switzerland; 61Department of Genomics, Life and Brain Center, Institute of Human Genetics, University of Bonn, Bonn, Germany; 62Institute of Neuroscience and Medicine, Research Center Jülich, Jülich, Germany; 63School of Psychology, Finders University, Adelaide, SA, Australia; 64Medical Research Council Social Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King's College London, London, UK; 65Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King's College London, London, UK; 66Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA; 67CHRU Montpellier, University of Montpellier, Montpellier, France; 68Department of Psychiatry, University of Minnesota, Minneapolis, MN, USA; 69Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 70MRC Integrative Epidemiology Unit, University of Bristol, Bristol, UK; 71Department of Psychosomatic Medicine and Psychotherapy, Hannover Medical School, Hannover, Germany; 72Department of Dietetics-Nutrition, Harokopio University, Athens, Greece; 73Department of Neurosciences, University of Padova, Padova, Italy; 74Seattle University College of Nursing, Seattle, WA, USA; 75Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA; 76First Department of Psychiatry, Athens University Medical School, Athens, Greece; 77University of Nantes, Nantes, France; 78Department of Chemistry and Biochemistry, University of California at San Diego, La Jolla, CA, USA; 79Laboratory of Psychiatric Genetics, Poznan University of Medical Sciences, Poznan, Poland; 80The Barcelona Institute of Science and Technology, Universitat Pompeu Fabra, CIBER Epidemiología y Salud Pública Barcelona, Barcelona, Spain; 81Interdisciplinary Cluster for Applied Genoproteomics-Université de Liège, Liège, Belgium; 82Stanford University, Stanford, CA, USA; 83Broad Institute of MIT and Harvard, Massachusetts General Hospital, MA, USA; 84Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, University Hospital of Würzburg, Würzburg, Germany; 85Division of Psychological and Social Medicine and Developmental Neurosciences, Faculty of Medicine, TU Dresden, Germany; 86Eating Disorders Research and Treatment Center, Department of Child and Adolescent Psychiatry, Faculty of Medicine, TU Dresden, Germany; 87The Barcelona Institute of Science and Technology, Universitat Pompeu Fabra, Centro de Investigación Biomédica en Red en Epidemiología y Salud Pública Barcelona, Barcelona, Spain; 88Estonian Genome Center, University of Tartu, Tartu, Estonia; 89Medical and Population Genetics Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA; 90Division of Endocrinology, Boston Children's Hospital, Boston, MA, USA; 91Department of Psychology, University of Oslo, Oslo, Norway; 92Centre for Genomic Regulation, The Barcelona Institute of Science and Technology, Universitat Pompeu Fabra, Centro de Investigación Biomédica en Red en Epidemiología y Salud Pública Barcelona, Barcelona, Spain; 93University Hospital of Bellvitge-IDIBELL, University of Barcelona, Barcelona, Spain; 94Centro de Investigación Biomédica en Red CIBEROBN, Instituto de Salud Carlos III, Madrid, Spain; 95Ludwig-Maximilians-University Munich, Schön Klinik Roseneck, Germany; 96UCL Institute of Cardiovascular Sciences, London, UK; 97Department of Cancer Epidemiology and Genetics, Masaryk Memorial Cancer Institute, Brno, Czech Republic; 98Clinical Genetics Unit, University Hospital of Padova, Padova, Italy; 99Lunenfeld-Tanenbaum Research Institute of Mount Sinai Hospital, University of Toronto, Toronto, ON, Canada; 100Ontario Institute for Cancer Research, Toronto, ON, Canada; 101Division of Nephrology and Dialysis Columbus-Gemelli University Hospital, Rome, Italy; 102Martin Luther University of Halle- Wittenberg, Halle, Germany; 103Eating Disorders Unit, First Department of Psychiatry, National and Kapodistrian University of Athens, Medical School, Athens, Greece; 104INSERM U984, Sainte- Anne Hospital, University of Paris-Descartes, Paris, France; 105Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, PA, USA; 106Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, PA, USA; 107The Division of Human Genetics, Department of Pediatrics, Perelman School of Medicine, Philadelphia, PA, USA; 108Department of Psychiatry, Weill Cornell Medical College, New York, NY, USA; 109Psychiatric Genetics Unit, Department of Psychiatry, Poznan University of Medical Sciences, Poznan, Poland; 110Department of Child and Adolescent Psychiatry and Psychotherapy, University Hospital Essen, University of Duisburg-Essen, Essen, Germany; 112Zorg op Orde, Leidschendam, The Netherlands; 113King’s College London, London, UK; 114Division of Medical Genetics, Department of Biomedicine, University of Basel, Basel, Switzerland; 115Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy of the RWTH Aachen, Aachen, Germany; 116Department of Psychosocial and Internal Medicine, Heidelberg University, Heidelberg, Germany; 117Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 118Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, UK; 119Icahn School of Medicine, Mount Sinai, NY, USA; 120Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, MA, USA; 121Eating Disorders Unit, Department of Child and Adolescent Psychiatry, Medical University of Vienna, Vienna, Austria; 122Department of Molecular Life Sciences, Tokai University School of Medicine, Kanagawa, Japan; 123Department of Epidemiology and Public Health, Ostrava University, Ostrava, Czech Republic; 124Department of Psychiatry and CIBEROBN, University Hospital of Bellvitge-IDIBELL, University of Barcelona, Barcelona, Spain; 125Department of Clinical Sciences, School of Medicine, University of Barcelona, Barcelona, Spain; 126Eating Recovery Center, Denver, CO, USA; 127Rheumatology Research Group, Vall d’Hebron Hospital Research Institute, Barcelona, Spain; 128Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; 129Social, Genetic and Developmental Psychology, King's College London, London, UK; 130Department of Psychiatry, First Faculty of Medicine, Charles University, Prague, Czech Republic; 131Department of Psychiatry, Center for Addiction and Mental Health, Institute of Medical Science, University of Toronto, Toronto, Canada; 132University of Helsinki, National Institute for Health and Welfare, Helsinki, Finland; 133Institute for Molecular Medicine Finland FIMM, University of Helsinki, Helsinki, Finland; 134Institute of Public Health and Clinical Nutrition, University of Eastern Finland, Kuopio, Finland; 135Groningen Institute for Evolutionary Life Sciences, University of Groningen, Groningen, The Netherlands; 136Brain Center Rudolf Magnus, Department of Translational Neuroscience, University Medical Center Utrecht, Utrecht, The Netherlands; 137Department of Psychiatry, University of California, San Diego, CA, USA; 138Center for Addiction and Mental Health, University of Toronto, Toronto, ON, Canada; 139Department of Public Health, Clinicum, University of Helsinki, Helsinki, Finland; 140Institute of Applied Health Sciences, University of Aberdeen, Aberdeen, UK; 141Department of Medicine, Unit of Rheumatology, Karolinska University Hospital, Stockholm, Sweden; 142Department of Psychology, Michigan State University, East Lansing, MI, USA; 143Health Data and Digitalisation, Norwegian Institute of Public Health, Oslo, Norway; 144Department of Medical Genetics, University Medical Center Utrecht, Utrecht, The Netherlands; 145Gothenburg University, Gothenburg, Sweden; 146NORMENT— K.G. Jebsen Center for Psychosis Research, Department of Clinical Science, University of Bergen, Bergen, Norway; 147Dr Einar Martens Research Group for Biological Psychiatry, Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway; 148Department of Psychiatry, University of Toronto, Toronto, ON, Canada; 149Department of Clinical Psychology, American School of Professional Psychology at Argosy University Washington DC, Washington, DC, USA; 150M. Sklodowska-Curie Cancer Center and Institute of Oncology, Warsaw, Poland; 151Department of Biological and Medical Psychology, University of Bergen, Bergen, Norway; 152K. G. Jebsen Center for Neuropsychiatric Disorders Norway, Oslo, Norway; 153Brain Mind Institute, EPFL, Center for Psychiatric Neuroscience, Department of Psychiatry—CHUV/UNIL, Lausanne, Switzerland; 154Division of Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia; 155Department of Psychiatry, University of Naples SUN, Naples, Italy; 156Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia; 157Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland; 158QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia; 159NORMENT, University of Oslo, Oslo, Norway; 160Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway; 161University College Cork, Health Service Executive South, Cork, Ireland; 162MRC Social Genetic and Developmental Psychiatry Centre, Kings College London, London, UK; 163Leiden University Medical Centre, Molecular Epidemiology Section (Department of Medical Statistics), Leiden, The Netherlands; 164Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 165Department of Psychiatry and Behavioral Science, University of North Dakota School of Medicine and Health Sciences, Fargo, ND, USA; 166National Center for PTSD, VA Boston Healthcare System, Boston, MA, USA; 167Boston University School of Medicine, Boston, MA, USA; 168Department of Medicine and Surgery, Section of Neurosciences, University of Salerno, Salerno, Italy; 169University of Naples SUN, Naples, Italy; 170Aarhus University, Denmark; 171Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden; 172Center for Molecular Medicine, Karolinska University Hospital, Stockholm, Sweden; 173Harakopio University, Athens, Greece; 174Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior, University of California Los Angeles, Los Angeles, CA, USA; 175Kartini Clinic, Portland, OR, USA; 176Institute for Molecular Medicine Finland, Helsinki, Finland; 177Centre de Psychiatrie et Neurosciences— Inserm U894, Paris, France; 178Department of Psychiatry, First Faculty of Medicine, Charles University, Prague, Czech Republic; 179Department of Psychiatry, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, Mount Sinai, NY, USA; 180University of Helsinki, Helsinki University Central Hospital, Helsinki, Finland; 181University of Turku, Turku, Finland; 182Department of Child and Adolescent Psychiatry, Poznan University of Medical Sciences, Poznan, Poland; 183Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK; 184Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Oxford, UK; 185Department of Genetics, Environment and Mental Health, Norwegian Institute of Public Health, University of Oslo, Oslo, Norway; 186Department of Biometry, University of Helsinki, Helsinki, Finland; 187Section of Eating Disorders, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, UK; 188University of Pisa, Pisa, Italy; 189Ludwig-Maximilians-University Munich, Munich, Germany; 190Institute of Psychiatry and Neurology, University of Social Sciences and Humanities, Warsaw, Poland; 191Research Group Clinical Epidemiology, Center for Sepsis Control and Care, Jena University Hospital, Jena, Germany; 192The Centre for Applied Genomics, Genetics and Genome Biology, The Hospital for Sick Children, Toronto, ON, Canada; 193McLaughlin Centre and Department of Molecular Genetics, University of Toronto, Toronto, ON, USA; 194Department of Human Biology, J. Craig Venter Institute, La Jolla, CA, USA; 195Department of Psychiatry and Psychotherapy, Medical University of Vienna, Vienna, Austria; 196Department of Pediatrics and Center of Applied Genomics, First Faculty of Medicine, Charles University, Prague, Czech Republic; 197McGill University and Génome Québec Innovation Centre, Montreal, QC, Canada; 198Leiden University Medical Centre, Leiden, The Netherlands; 199Rivierduinen Eating Disorders Ursula, Leiden University Medical Centre, Leiden, The Netherlands; 200Department of Psychiatry, Poznan University of Medical Sciences, Poznan, Poland; 201Department of Human Genetics, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, UK; 202Department of Haematology, University of Cambridge, Hills Rd, Cambridge CB2 0AH, UK; 203The National Institute for Health Research Blood and Transplant Unit (NIHR BTRU) in Donor Health and Genomics at the University of Cambridge, Cambridge, UK; 204Department of Clinical Science, K.G. Jebsen Centre for Psychosis Research, Norwegian Centre For Mental Disorders Research, University of Bergen, Bergen, Norway; 205Dr Einar Martens Research Group for Biological Psychiatry, Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway; 206Department of Pathology, University Medical Center Utrecht, Utrecht, The Netherlands; 207Department of Psychiatry and Biobehavioral Sciences, University of California Los Angeles, Los Angeles, CA, USA; 208David Geffen School of Medicine, University of California, Los Angeles, CA, USA; 209Department of Genetics and Psychiatry, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 210Nofer Institute of Occupational Medicine, Department of Environmental Epidemiology, Lodz, Poland; 211University of Padova, Padova, Italy; 212University of Perugia, Perugia, Italy; 213eHealth Lab-Computer Science Department, University of Cyprus, Nicosia, Cyprus; 214Adolescent Health Unit (A.H.U.), 2nd Department of Pediatrics—Medical School, University of Athens "P. & A. Kyriakou" Children's Hospital, Athens, Greece; 215"P. & A. Kyriakou" Children's Hospital, Athens, Greece; 216Center for Eating Disorders Rintveld, Altrecht, The Netherlands; 217Rivierduinen Eating Disorders Ursula, Leiden, The Netherlands; 218Department of Psychiatry, Leiden University Medical Center, Leiden, The Netherlands; 219Department of Psychology, Georgia State University, Atlanta, GA, USA; 220Department of Psychology, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, UK; 221University of North Carolina at Chapel Hill, School of Paediatrics and Child Health, Faculty of Medicine, Dentistry, and Life Sciences, The University of Western Australia, Perth, WA, Australia; 222School of Psychology and Speech Pathology, Faculty of Health Sciences, Curtin University, Perth, WA, Australia; 223Helmholtz Centre Munich—German Research Center for Environmental Health, Munich, Germany; 224University of Toronto, Toronto, ON, Canada; 225Inpatient Eating Disorders Service, Toronto General Hospital, Toronto, ON, Canada; 226Section on Growth and Obesity, Program in Developmental Endocrinology and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA and 227Department of Internal Medicine VI, Psychosomatic Medicine and Psychotherapy, University Medical Hospital Tuebingen, Tuebingen, Germany
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Huckins, L., Hatzikotoulas, K., Southam, L. et al. Investigation of common, low-frequency and rare genome-wide variation in anorexia nervosa. Mol Psychiatry 23, 1169–1180 (2018). https://doi.org/10.1038/mp.2017.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.88
This article is cited by
-
Differential methylation patterns in lean and obese non-alcoholic steatohepatitis-associated hepatocellular carcinoma
BMC Cancer (2022)
-
What next for eating disorder genetics? Replacing myths with facts to sharpen our understanding
Molecular Psychiatry (2022)
-
Genome-wide association study reveals new insights into the heritability and genetic correlates of developmental dyslexia
Molecular Psychiatry (2021)
-
SIRT1 accelerates the progression of activity-based anorexia
Nature Communications (2020)
-
Histone deacetylase 4 (HDAC4): a new player in anorexia nervosa?
Molecular Psychiatry (2019)
