
Abstract
Obsessive-compulsive disorder (OCD) is a common neuropsychiatric disease affecting about 2% of the general population. It is characterized by persistent intrusive thoughts and repetitive ritualized behaviors. While gene variations, malfunction of cortico-striato-thalamo-cortical (CSTC) circuits, and dysregulated synaptic transmission have been implicated in the pathogenesis of OCD, the underlying mechanisms remain largely unknown. Here we show that OCD-like behavior in mice is caused by deficiency of SPRED2, a protein expressed in various brain regions and a potent inhibitor of Ras/ERK-MAPK signaling. Excessive self-grooming, reflecting OCD-like behavior in rodents, resulted in facial skin lesions in SPRED2 knockout (KO) mice. This was alleviated by treatment with the selective serotonin reuptake inhibitor fluoxetine. In addition to the previously suggested involvement of cortico-striatal circuits, electrophysiological measurements revealed altered transmission at thalamo-amygdala synapses and morphological differences in lateral amygdala neurons of SPRED2 KO mice. Changes in synaptic function were accompanied by dysregulated expression of various pre- and postsynaptic proteins in the amygdala. This was a result of altered gene transcription and triggered upstream by upregulated tropomyosin receptor kinase B (TrkB)/ERK-MAPK signaling in the amygdala of SPRED2 KO mice. Pathway overactivation was mediated by increased activity of TrkB, Ras, and ERK as a specific result of SPRED2 deficiency and not elicited by elevated brain-derived neurotrophic factor levels. Using the MEK inhibitor selumetinib, we suppressed TrkB/ERK-MAPK pathway activity in vivo and reduced OCD-like grooming in SPRED2 KO mice. Altogether, this study identifies SPRED2 as a promising new regulator, TrkB/ERK-MAPK signaling as a novel mediating mechanism, and thalamo-amygdala synapses as critical circuitry involved in the pathogenesis of OCD.
Similar content being viewed by others

Introduction
Obsessive-compulsive disorder (OCD) is a neuropsychiatric condition characterized by persistent intrusive thoughts (obsessions) and repetitive ritualized actions (compulsions). Factor analytic studies have identified four primary subtypes of OCD: contamination obsessions with cleaning compulsions, symmetry obsessions with ordering compulsions, hoarding obsessions with collecting compulsions, and aggressive/sexual/religious/somatic obsessions with checking compulsions.1 However, OCDs vary greatly in the types of obsessions and compulsions, reflecting both heterogeneity in clinical phenotypes and the underlying pathophysiology.2, 3 Furthermore, there are various OCD-related disorders, for example trichotillomania and excoriation disorder, tic disorders like Tourette’s syndrome, and autism spectrum disorders that share considerable overlapping features with OCD.4
As with many neuropsychiatric disorders, the neurobiological basis of OCD still remains obscure. A large body of functional neuroimaging studies has related OCD symptoms to alterations in the activity of cortico-striato-thalamo-cortical (CSTC) circuits.5, 6 Especially hyperactivity in orbitofrontal cortex and ventromedial striatum seems to be crucial in the pathogenesis of OCD.7 The amygdala is the integrative center for emotions and emotional behavior and its role in mediating fear and anxiety is the most commonly referenced to date.8, 9 However, a possible impact of the amygdala on the development of OCDs is indicated and extensively discussed but needs additional investigation.10
Although family and twin studies support a significant genetic contribution to OCD and related conditions, no particular gene has reached the stringent level of statistical evidence to be considered a definitive risk gene.4 Since selective serotonin reuptake inhibitors (SSRIs), such as fluoxetine, are the first-line pharmacological treatment for OCDs, one of the strongest candidates for the cause of OCD is the gene encoding the serotonin transporter.11 The same meta-analysis also implicated glutamatergic and dopaminergic neurotransmitter systems as well as brain-derived neurotrophic factor (BDNF) and tropomyosin receptor kinase B/neurotrophic tyrosine kinase receptor type 2 (TrkB/NTRK2) as possible genetic factors.11 The latter two are part of a cerebral signaling pathway, which is essential for the regulation of neuronal gene transcription, neurogenesis, and neuronal differentiation. In the adult nervous system, BDNF/TrkB signaling regulates synaptic strength, transmission, and plasticity.12, 13 Because BDNF plays a critical role in brain development and plasticity, it is widely implicated in psychiatric diseases, including major depressive, bipolar, anxiety-related, and neurodevelopmental disorders but also in neurodegenerative diseases.14, 15 Although several studies demonstrated the involvement of BNDF in OCD as well, the outcomes were inconclusive in a way that it is still unclear whether BDNF sequence variants like the common Val66Met substitution are protective or predictive for OCD.16, 17 Alterations in BDNF plasma levels are indicative of various psychiatric disorders14 and may also be associated with OCD.18 Genetic variations of the NTRK2 gene encoding TrkB were suggested to contribute to OCD in humans, however, the pathomechanism is unknown.19 The impact of BDNF and its receptor TrkB on anxiety-related disorders has been investigated in mouse models but this also revealed contradictory results.20
The neurotrophin BDNF is the most prevalent growth factor in the central nervous system and the preferred ligand of TrkB, a transmembrane receptor tyrosine kinase that is phosphorylated at several tyrosine residues.21 After BDNF-mediated activation of TrkB, signals are mediated by different intracellular cascades, of which the Ras/ERK-MAPK pathway is one of the most prominent.12, 13 Ras is activated after receptor phosphorylation via different adapter molecules. From Ras, signals can be propagated by phosphorylation and activation of sequential kinases including Raf, MEK1/2, and ERK1/2. ERK activation results in phosphorylation and activation of transcription factors and other regulatory target proteins.22 Important intrinsic regulators of Ras/ERK-MAPK pathway activity are the SPRED proteins (Sprouty-related, EVH1 domain-containing protein 2), a family of the three homologs SPRED1, 2 and 3.23 SPREDs very potently and exclusively inhibit the Ras/ERK-MAPK cascade downstream of multiple receptor tyrosine kinases and in response to a wide range of mitogenic stimuli, for example, growth factors, cytokines, and chemokines.23, 24, 25 The expression pattern of SPRED proteins, especially that of SPRED2 is widespread in humans and mice but most pronounced in the central nervous system.26, 27 SPREDs are also functionally required in neuronal development by regulating neurogenesis through control of ERK-dependent neural progenitor cell proliferation and maintenance of germinal zone integrity.28 Because of the co-localization of SPREDs with various endosome markers, a role in synaptic vesicle transport is also assumed.27, 28, 29 SPRED1 deficiency in humans causes Legius syndrome, a disease of the rasopathy spectrum, which is associated with learning disabilities, developmental delays, and macrocephaly.30 Similarly, SPRED1 KO mice also show disabilities in hippocampus-dependent learning31 and facial abnormalities.32 The loss of functional SPRED2 in mice leads to hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis, demonstrated by increased release of stress hormones including corticotropin-releasing hormone, adrenocorticotropic hormone, and corticosterone.33
Here we show that SPRED2 deficiency caused an OCD-like behavioral phenotype. The observation of OCD-like grooming resulting in severe self-inflicted facial lesions in SPRED2 KO mice prompted us to investigate anxiety-related behavior and skin sensitivity, and to treat SPRED2 KO mice with fluoxetine. Since our results supported the assumption of an OCD-like phenotype in SPRED2 KO mice, we performed electrophysiological recordings at cortico-striatal and thalamo-amygdala synapses in SPRED2 KOs. We recorded distinct changes of synaptic excitability at thalamo-amygdala synapses, which was accompanied by altered neuron morphology in the lateral amygdala. Hence, we aimed to unravel the cause of thalamo-amygdala malfunction and to verify its contribution to OCD. Investigation of SPRED2 expression and of various pre- and postsynaptic proteins in wildtype (WT) and SPRED2 KO amygdala identified the molecular basis for dysregulated activity in thalamo-amygdala circuits. Given the described inhibitory effect of SPRED2 on Ras/ERK-MAPK signaling, which is an essential mediator of BDNF/TrkB signals in brain, we considered TrkB/ERK pathway dysregulation as a cause of synaptic protein level alterations in the amygdala. We investigated expression and activity of crucial signaling components like BDNF, TrkB, Ras and ERK and demonstrated that pathway upregulation is a specific result of SPRED2 deficiency. Our hypothesis that TrkB/ERK-MAPK pathway overactivation contributes to OCD was confirmed by artificial pathway downregulation using selumetinib, which restored normal behavior in SPRED2 KO mice. With this study, we discovered a link between SPRED2 deficiency, TrkB/ERK signaling, thalamo-amygdala malfunction, and psychiatric conditions like OCD.
Materials and methods
SPRED2 KO mice
SPRED2 KO mice were generated by a gene trap approach as described previously.34, 35 To generate mice with a disrupted, non-functional Spred2 gene, the embryonic stem cell line XB228 (International Gene Trap Consortium, Davis, CA, USA) was used. It contained the pGT0 gene trap vector, which functionally disrupted the Spred2 gene (Figure 1b). Mice were housed in a daily 12/12 h light-dark cycle under controlled room temperature (21±1 °C) and humidity (55±5%) conditions with tap water and standard mouse chow ad libitum unless stated otherwise. To minimize possible inbred effects, mice were raised on a mixed 129/Ola × C57Bl/6 genetic background. SPRED2 KO mice were obtained by mating SPRED2 heterozygous animals. All mouse experiments were conducted using SPRED2 KO mice and WT littermates as controls. Unless stated otherwise, we used SPRED2 KO mice aged 6–12 months with apparent OCD-like phenotype of mixed gender. Mice too severely affected by their behavior and thereby not suitable for testing were excluded from the analyses. Required sample sizes were calculated based on effect size and error probabilities using G*Power 3.1.9.2.36 Numbers of samples (n) used in each experiment are indicated in figure legends. All experiments were approved by the local councils for animal care (Regierung von Unterfranken: #98/14; #03/12; #2-375) and were conducted according to the European law for animal care and use.

Self-inflicted facial lesions result from OCD-like grooming in SPRED2 KO mice. (a) Severe facial skin lesions developed mostly uni- and sometimes bilaterally in SPRED2 KO mice from the age of 4 months. These lesions were a result of excessive and injurious self-grooming, appeared on head, neck and snout regions and were characterized by ulcerations and hemorrhage. (b) SPRED2 KO mice were generated by insertion of a gene trap vector between exons 4 and 5 of Spred2. Gene trapping resulted in a non-functional Spred2 gene and the Spred2 promoter-driven expression of the β-geo reporter gene. (c) In vivo monitoring of Spred2 expression by X-Gal staining of coronal brain sections of SPRED2 heterozygous mice demonstrated widespread SPRED2 expression in the brain including cortex, hippocampus, amygdala and striatum, the latter two brain regions associated with the development of OCDs. (d) Exemplary Western blot analyses revealed SPRED2 expression in WT amygdala lysates and the complete absence of SPRED2 protein in amygdala of SPRED2 KO mice. Expression of related SPRED1 was not compensatory upregulated as confirmed by quantification of relative SPRED1 expression (n=11 for WT and KO). Data are mean±s.e.m. KO, knockout; n.s., not significant; OCD, obsessive-compulsive disorder; WT, wildtype.
Behavioral analysis
Open field
The open field (OF) consisted of a quadratic black opaque PERSPEX XT box (50 × 50 × 40 cm), which was semipermeable to infrared light (TSE Systems, Bad Homburg, Germany) and illuminated by infrared LEDs from below.37 The area of the OF was divided into a 36 × 36 cm central zone (100 lx) and the surrounding periphery (50 lx). Mice were placed in the periphery and their behavior was recorded for 5 min using the VideoMot2 system (TSE Systems). Variables measured included time spent, distance traveled, and visits in each zone, total time spent moving, total distance traveled, vertical rears, number of grooming bouts, and defecation/urination.
Elevated plus maze
An elevated plus maze (EPM) made from black PERSPEX (TSE Systems) and semipermeable for infrared light was used and illuminated by infrared LEDs from below.37 The apparatus was elevated to a height of 60 cm above floor level and comprised a central platform (5 × 5 cm, 15 lx) extending to two opposing open arms (30 × 5 × 0.25 cm, 30 lx) and two opposing closed arms (30 × 5 × 15 cm, 5 lx). Mice were placed in the center facing an open arm, and their behavior was recorded for 5 min using the VideoMot2 system (TSE Systems). Behavioral analysis included time spent, distance traveled, and visits in each zone, total distance traveled, total time spent moving, vertical rears, number of grooming bouts, and defecation/urination.
Light/dark box
The light/dark box (LDB) contained a central gate (5 × 5 cm) separating a transparent, brightly illuminated 'lit' compartment (40 × 40 × 27 cm, 300 lx) from a small enclosed 'dark' compartment (40 × 20 × 27 cm, 0–5 lx).38 Mice were placed into the light compartment and their behavior was recorded for 5 min. Measured behavioral parameters included time spent in each compartment, latency to cross from lit to dark area, number of grooming bouts, and defecation/urination.
Mechanical sensitivity
The von Frey test based on the up-and-down-method39 was used to examine paw withdrawal thresholds to mechanical stimulation. Mice were placed in plexiglass cages on a wire mesh and the plantar surface of the hindpaws was touched with a von Frey filament starting at 0.69 g. If the mouse withdrew its hindpaw upon administration of mild pressure, the next thinner von Frey filament was used. If the mouse showed no reaction to this stimulation, the next thicker von Frey filament was applied. Each hindpaw was tested three times. The 50% withdrawal threshold (that is force of the von Frey hair to which an animal reacts in 50% of the administrations) was recorded. Tests were performed by an investigator blinded for mouse genotype and study objectives.
Thermal sensitivity
Paw withdrawal latencies to heat were determined applying a standard algesiometer (Ugo Basile, Gemonio, Italy) based on the method of Hargreaves.40 Mice were placed on a glass surface and a radiant heat source was positioned under one hindpaw. The time until paw withdrawal was recorded automatically. To avoid tissue damage, a time limit for heat application of 15 s was used. Each hindpaw was tested three times. Tests were performed by an investigator blinded for mouse genotype and study objectives.
Grooming behavior
Grooming included face-wiping, full-body grooming, and scratching and rubbing of head and ears. Grooming bouts lasted at least 3 s; bouts after pauses longer than 3 s were regarded as new bouts. Number of grooming bouts were counted during EPM, OF and LDB tests. Mouse behavior was also determined by video observations before and after experimental rescue with fluoxetine. Mice were placed in a standard cage, which was covered by a closed chamber containing a webcam (Philips, Amsterdam, Netherlands) and infrared LEDs for illumination on top. Every mouse was recorded for 30 min and movies were analyzed for the times mice spent with grooming, digging, rearing, locomotion and the distance traveled using VideoMot2 (TSE Systems).
Experimental therapy
Fluoxetine treatment
SPRED2 KO mice and WT littermate controls were treated with the SSRI fluoxetine (Stada, Bad Vilbel, Germany) for 2 weeks. Mice of each genotype were randomly selected for either the placebo or fluoxetine group. Fluoxetine was administered at a dose of 20 mg kg−1 day−1 within standard mouse diet; the placebo group was fed with standard mouse diet. Documentation by photos (Canon EOS 1000D digital camera, Tokyo, Japan) and videos (Philips PixelPlus webcam) was performed before and after 2 weeks of treatment.
Selumetinib treatment
SPRED2 KO mice were treated with the MEK1/2 inhibitor selumetinib (AZD6244, Selleck chemicals, Houston, TX, USA) for one week. Selumetinib was administered at a dose of 8 mg kg−1 day−1 within standard mouse diet. Photo documentations were performed with a Canon EOS 1000D digital camera before and after one week of treatment.
Electrophysiology
Mice were terminally anesthetized with isoflurane, decapitated, brains were rapidly removed and transferred into ice-cold preparation solution containing (in mM): 210 sucrose, 26 NaHCO3, 1.3 MgSO4, 1.2 KH2PO4, 2 MgCl2, 2 KCl, 2 CaCl2, 10 glucose, 3 myoinositol, 2 sodium-pyruvate, and 0.4 ascorbic acid, equilibrated with 95% O2/5% CO2. Coronal slices (250 μm) were cut with a vibratome (Leica VT1000S, Wetzlar, Germany) in a submerged chamber filled with ice-cold preparation solution. Slices were transferred to a holding chamber filled with artificial cerebrospinal fluid containing (in mM): 124 NaCl, 26 NaHCO3, 2 KCl, 1.2 KH2PO4, 1.3 MgSO4, 2 CaCl2, and 10 glucose, equilibrated with 95% O2/5% CO2. The holding chamber was heated for 1 h to 34 °C to improve patch success before slices were kept at room temperature. For recording, slices were transferred into a superfusion recording chamber mounted on an upright fixed stage microscope (Zeiss, Oberkochen, Germany) with infrared differential interference optics. Superfusion rate was 2-3 ml artificial cerebrospinal fluid per minute. Patch-clamp recordings were made at room temperature under visual guidance by an infrared sensitive camera (Kappa CF6, Gleichen, Germany). Patch electrodes were pulled from borosilicate capillaries (Science Products, Hofheim, Germany) and filled with a solution containing (in mM): 95 K-gluconate, 20 K3-citrate, 10 NaCl, 1 MgCl2, 0.5 CaCl2, 1 BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid), and 10 HEPES, pH 7.2 (KOH), 270–290 mosm/l. Measurements of miniature excitatory postsynaptic currents (mEPSCs) were made with a high chloride solution containing (in mM): 140 KCl, 5 NaCl, 1 CaCl2, 10 EGTA, 2 MgCl2, 2 K2-ATP, 0.5 Na-GTP, and 10 HEPES, pH 7.2 (KOH), 270–290 mosm/l. Electrodes had a resistance of 2–4 MΩ.
Synaptic input to lateral amygdala and putamen neurons was investigated by placing a SNEX-200 concentric tungsten electrode (Science Products, Hofheim, Germany) in the afferent fiber tract. The electrode was connected to a stimulator (ISO-Flex, A.M.P.I., Jerusalem, Israel). As marked in Figure 3, appropriate brain regions were identified according to the mouse brain atlas by Paxinos and Franklin.41 During whole cell recordings a presynaptic stimulus was applied in voltage-clamp configuration at −70 mV for 150 μs. Amplitudes of 10 responses were always averaged (1 s repeat interval). The stimulation threshold was the minimal stimulation amplitude (in mA) eliciting a postsynaptic response. Recordings were made using an EPC10 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Data were filtered with a 10 kHz Bessel and 2.9 low-pass Bessel filter with a sampling rate of 4–30 kHz. The Pulse and Pulsefit software (HEKA Elektronik) was used for data acquisition and analysis. mEPSC were measured in voltage clamp configuration (−70 mV) for 1 min with 200 μM CdCl2 in artificial cerebrospinal fluid. For data analysis we used Mini Analysis 6.0 software (Synaptosoft, Decatur, GA, USA). Resistance and seal quality were monitored at the beginning and several times during recordings to assure consistent measurement conditions.

Altered synaptic excitability in amygdala and striatum of SPRED2 KO mice. (a) Schematic brain slice showing representative positions of afferent fiber stimulation (asterisk) in the corpus callosum and of whole cell current measurements in the putamen. The location of each measured cell is marked by a circle. (b) Exemplary excitatory postsynaptic currents (EPSC1, EPSC2) for SPRED2 KO and WT recorded after two consecutive afferent fiber stimulations (asterisks). (c) Stimulation threshold in putamen of SPRED2 KOs tended to be reduced after stimulation of afferent corpus callosum fibers. (d) Both EPSC1 and EPSC2 were elevated comparably in putamen of SPRED2 KO mice after two consecutive afferent fiber stimulations (paired pulses). (e) Paired pulse ratio of WTs and KOs was similar in putamen. n=5 for WT and SPRED2 KO in each experimental setup of cortico-striatal neurotransmission measurement. (f) Schematic brain slice showing representative positions of thalamic afferent fiber stimulation (asterisk) and of whole cell current measurements in the lateral amygdala. (g) Exemplary excitatory postsynaptic currents (EPSC1, EPSC2) for SPRED2 KO and WT recorded after two consecutive afferent fiber stimulations (asterisks). (h) Stimulation threshold in lateral amygdala of SPRED2 KOs was decreased after stimulation of afferent thalamic fibers. (i) In comparison to WTs, consecutive afferent fiber stimulations (paired pulses) revealed an increased EPSC1 but a similar EPSC2 in lateral amygdala of SPRED2 KO mice. (j) Accordingly, the paired pulse ratio was reduced in lateral amygdala of SPRED2 KOs. (k) Exemplary miniature excitatory postsynaptic currents (mEPSCs) measured in lateral amygdala of WT and SPRED2 KO cells. (l) mEPSC frequency was decreased in SPRED2 KO mice. (m) mEPSC magnitude was reduced in SPRED2 KOs. n=8 for WT and n=9 for SPRED2 KO in each experimental setup of thalamo-amygdala neurotransmission measurement. Data are mean±s.e.m; *P<0.05. KO, knockout; n.s., not significant; WT, wildtype.
Neuron morphology
Mouse neurons were stained using a modified Golgi-Cox impregnation method as described previously.42 In brief, dissected brains were impregnated in Golgi solution for 30 days, 150 μm serial coronal slices were prepared using a sliding microtome and mounted on glass microscopic slides using Vitro Clud (R. Langenbrinck, Emmendingen, Germany). Golgi-stained pyramidal neurons in the lateral amygdala were reconstructed by an experimenter blind to the genotype using the Neurolucida system (MBF Bioscience, Williston, VT, USA). Only neurons located in the center of sections, displaying intense staining of dendritic arborizations and allowing unequivocal identification of dendritic spines were chosen for reconstructions. Per genotype 20–30 neurons were used to determine dendritic parameters such as length of dendrites, spine numbers, and spine densities/10 μm in total or of a particular branch order.
Quantitative real-time PCR
Amygdala was punched out from mouse brains, tissues were homogenized with a Polytron PT 3100 homogenizer (Kinematica, Luzern, Switzerland) and RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). 500 ng of total RNA were reverse transcribed to complementary DNA (cDNA) using TaqMan Reverse Transcription Reagents (Applied Biosystems, Waltham, MA, USA) in a 100 μl PCR reaction additionally containing: 10 × Reaction Buffer (10 μl), 10 mM dNTPs (20 μl), 25 mM MgCl2 (22 μl), Random Hexameres (5 μl), RNAse Inhibitor (2 μl) and 50 U/μl Multiscribe Reverse Transcriptase (6.25 μl). The 96-well GeneAmp PCR System 9700 cycler was used and the following cycler conditions obtained: 10 min, 38 °C; 60 min, 48 °C; 25 min, 95 °C. Five μl of cDNA entered quantitative real-time PCR (qRT-PCR) using TaqMan Universal Master Mix and the following target specific predesigned mouse TaqMan Gene Expression Assays (Applied Biosystems; Assay-IDs in brackets): PSD 95 (Mm00492193_m1), mGluR2 (Mm01235831_m1), mGluR5 (Mm00690332_m1), and ERC1 (Mm00453569_m1). 18 s rRNA (Hs99999901_s1) was used as an endogenous control. The 2−ΔΔCq method was applied for relative quantification of gene expression as previously described.43 Mean value of WT samples was set to 1 and mean value of KO samples was expressed as x-fold of WT.
Preparation of amygdala lysates
Amygdala was dissected from mouse brains on a metal plate cooled with ice and homogenized in an assay-dependent buffer using a plastic douncer fitting into a 1.5 ml microreaction tube. Protein content of samples was measured using the Bradford method.
Western blot
Amygdala lysates were prepared by adding 1 ml of 2% SDS in PBS supplemented with Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche) to 50 mg of tissue. Proteins were separated by 5–15% SDS–PAGE under reducing conditions and electrotransferred to Protran nitrocellulose membranes using semi-dry blotters. Blots were probed using primary antibodies against GAPDH (#2118, 1:10,000, Cell Signaling Technology, Danvers, MA, USA), SPRED1 (1:500,34), SPRED2 (1:500,34), PSD95 (#P246, 1:2,000, Sigma-Aldrich, St Louis, MO, USA), mGluR5 (#53090, 1:500, Abcam, Cambridge, UK), mGluR2 (#15672, 1:1,000, Abcam), ERC1 (#50312, 1:500, Abcam), Bassoon (# 141003, 1:500, Synaptic Systems, Goettingen, Germany), Rab3A (#107111, 1:1,000, Synaptic Systems), Rab6 (#sc310, 1:200, Santa Cruz Biotechnology, Dallas TX, USA), α-Tubulin (#T6074, 1:5,000, Sigma-Aldrich), β-Tubulin (#T8320, 1:2,000, Sigma-Aldrich), p44/42 MAP kinase (#9102, ERK1/2, 1:2,000, Cell Signaling Technology), phospho-p44/42 MAP kinase (P-ERK1/2, #9101, 1:1,000, Cell Signaling Technology), TrkB (#07225, 1:1,000, Merck Millipore, Billerica, MA, USA), phospho-TrkB Y515 (P-TrkB Y515, #ab109684, 1:150, Abcam) and phospho-TrkB Y817 (P-TrkB Y817, #bs-3732R, 1:200, Bioss, Woburn, MA, USA) followed by goat anti-rabbit (#111-035-144, 1:10,000, Jackson Immuno Research, West Grove, PA, USA) or goat anti-mouse (#115-035-146, 1:5,000, Jackson Immuno Research) horseradish peroxidase-conjugated secondary antibodies, all diluted in 5% non-fat dry milk in PBS supplemented with 0.05% Tween20. Signals were developed using CheLuminate-horseradish peroxidase FemtoDetect reagent (Applichem, Darmstadt, Germany) and recorded by a FluorChem SP Imager (Alpha Innotech, Biozym, Hessisch Oldendorf, Germany).
Ras activity assay
A pan-Ras Activation Assay Kit (#STA-400, Cell Biolabs, San Diego, CA, USA) was used to detect active Ras in mouse amygdala lysates according to the manufacturer’s protocol. Tissue was lysed at a ratio of 50 mg per 1 ml assay buffer and 1 mg total protein was used for each Ras pull-down.
Quantification of protein expression
Band intensities of Western blots were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Expression levels of proteins were normalized to GAPDH expression, P-ERK levels were normalized to total ERK, P-TrkB levels to total TrkB, and levels of active Ras to total Ras signals. Mean value of WT samples was set to 1 and mean value of KO samples was expressed as x-fold of WT.
Phospho-RTK array
Active phosphorylated receptor tyrosine kinases (RTK) in amygdala lysates were identified using the membrane-based Proteome Profiler Mouse Phospho-RTK Array (#ARY014, R&D Systems, Minneapolis, MN, USA). For incubation of each membrane 500 μg of protein was used, following the instructions of the manufacturer's protocol. Signals were detected by a FluorChem SP Imager (Alpha Innotech, Biozym).
BDNF ELISA
For determination of mature free BDNF levels in mouse amygdala lysates, we used the BDNF Emax ImmunoAssay System (#G7610, Promega, Madison, WI, USA) without an acid treatment procedure according to the manufacturer’s instructions. All samples were analyzed in duplicate, BDNF concentrations were calculated from a standard curve using nonlinear regression curve fitting (Prism5, GraphPad, La Jolla, CA, USA) and related to the corresponding protein content.
SPRED2 phosphorylation assay
Embryonic mouse hypothalamic cells (cell line mHypoE-44, Cedarlane, Burlington, ON, Canada) were cultivated in high glucose DMEM (Invitrogen), supplemented with 10% FBS and 1% Pen/Strep at 37 °C and 5% CO2 and grown to 90–100% confluency. To starve for growth factors, cells were rinsed twice with sterile PBS and incubated at 37 °C and 5% CO2 in DMEM without FBS. After 6 h, cells were stimulated with 50 ng ml−1 BDNF in DMEM (Merck Millipore) for 5, 15, 30 or 60 min. Cells were lysed in 1 ml of ice-cold buffer containing 150 mM NaCl, 1% Igepal CA-630, 0.5% Sodium deoxycholate, Complete Protease Inhibitor Cocktail (Roche) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche). Lysates were centrifuged for 10 min at 12 000g and 4 °C and supernatants were collected. As an input control 50 μl of each sample was mixed with 50 μl Laemmli buffer and incubated for 5 min at 95 °C. For antibody incubation, 5 μg phospho-tyrosine (PY) antibody mix containing 3 μg anti-PY 100 and 2 μg anti-PY 102 (#9411 and #9416, Cell Signaling Technology) was added to each sample before rotating for 1 h at 4 °C. Precipitation of the antibody-protein complexes was performed by adding 100 μl of 50% v/v Protein G Sepharose 4 Fast Flow beads (GE Healthcare, Chalfont St Giles, UK) to protein samples and incubating for 1 h at 4 °C under gentle rotation. After washing of beads 2 times with 1 ml lysis buffer and once with 1 ml 50 mM Tris buffer (pH 8.0) they were mixed with 50 μl Laemmli buffer and incubated for 5 min at 450 r.p.m. and 95 °C. Samples were stored at −20 °C and analyzed by Western blot.
X-Gal staining
Dissected mouse brains were embedded in Tissue-Tek OCT-compound (Sakura Finetek Europe, Leiden, Netherlands) and snap-frozen in liquid nitrogen. 10 μm cryosections were cut using a CM1950 microtome (Leica) and X-Gal staining was performed as previously described.26, 35 Stained sections were photographed by a Canon EOS 1000D digital camera connected to a Stereomicroscope 2000-C (Zeiss).
Statistics
Data obtained from the fluoxetine rescue experiment were first analyzed via three-way mixed ANOVA with genotype (WT/KO) and treatment (fluoxetine/placebo) as between-subjects factors and time (baseline/2 weeks) as repeated measures factor. Post hoc t-tests were performed to further analyze significant time × treatment × genotype interactions. In the remaining experiments, significant differences between genotypes were analyzed by Mann–Whitney U test for not normal distributed data or by two-sided two-sample t-tests and Welch's tests, respectively, depending on the homogeneity of variances. Normal distribution of data was tested using the Shapiro–Wilk test in combination with graphical analysis tools like box and histogram plots. Statistical analyses were performed using SPSS Statistics 19 (IBM, Armonk, NY, USA) or Prism5 (GraphPad). Results are expressed as mean±s.e.m. A P-value of *P<0.05 was considered statistically significant, whereas **P<0.01 represented high and ***P<0.001 highest significance. P-values⩾0.05 were considered as statistically not significant (n.s.).
Results
Self-inflicted facial lesions of SPRED2 KO mice are indicative of obsessive-compulsive grooming
Starting at ~4 months of age, SPRED2 KO mice developed apparent skin lesions on head, neck, and snout regions. These lesions occurred uni- and bilaterally and progressed to ulcerations with hemorrhage over time (Figure 1a). The penetrance of this phenotype increased with age, and 80% of the KOs were affected at the age of 12 months. We did not detect any lesions in WT littermates, even when they were housed in the same cage with SPRED2 KO mice from birth. This indicated that the lesions in KO mice were not a result of aggressive encounters between cage mates. However, SPRED2 KO mice were often seen engaged in self-grooming regardless of whether they were housed alone or with littermates (Supplementary Video). Thus, we hypothesized that the phenotype of SPRED2 KOs could be the result of excessive and injurious self-grooming, indicating an OCD-like behavior caused by SPRED2 deficiency in brain regions relevant for the onset of OCD.
In SPRED2 KO mice, the Spred2 gene was disrupted by insertion of a gene trap vector between exons 4 and 5 of Spred2 (Figure 1b). The gene trap vector comprised a β-geo reporter gene, which is expressed under control of the endogenous Spred2 promoter. Therefore, in vivo monitoring of Spred2 expression by X-Gal staining is possible. Indicated by the blue color after X-Gal staining of coronal brain sections from heterozygous mice, SPRED2 is expressed in various regions of the brain, including cortex and hippocampus. High promoter activity was especially detected in amygdala and striatum, both brain regions associated with the development of OCD-like behaviors (Figure 1c). Western blot analysis using amygdala lysates from SPRED2 KO mice and WT controls demonstrated SPRED2 expression in WT amygdala but the complete loss of full-length SPRED2 protein in KO amygdala. The deficiency of functional SPRED2 was not compensated by increased expression of homologous SPRED1, demonstrated by unaltered SPRED1 expression after normalization to GAPDH (Figure 1d).
Obsessive-compulsive grooming is associated with changes in anxiety-like behavior in SPRED2 KO mice
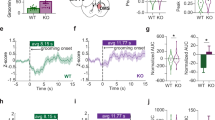
Excessive grooming or other OCD-related conditions are often associated with additional behavioral phenotypes in mice. To assess anxiety-like behavior in SPRED2 KO mice, we performed OF, EPM and LDB tests. We used male mice aged 7–10 months, displaying excessive grooming and facial lesions. In the OF an elevated number of grooming events was recorded in SPRED2 KO mice (Figure 2a), supporting our hypothesis of an obsessive-compulsive behavior as cause of the self-inflicted skin lesions. The total distance traveled was reduced in SPRED2 KO mice, indicating diminished locomotor and exploratory activity as a consequence of obsessive grooming (Figure 2a). In comparison to WTs, SPRED2 KO mice tended to spend more time in the center of the OF (Supplementary Table 1), which was indicative of less anxiety. In the EPM, SPRED2 KOs in fact spent a longer time span in the open arms (Figure 2b), traveled a longer distance in, and payed more visits to the open area as compared with control mice; the parameters in the guarded area were accordingly decreased (Supplementary Table 1). Normally, mice avoid exploration of the potentially dangerous open arms, which pointed to reduced anxiety in SPRED2 KO mice. Similar to the OF test, the total distance traveled during the EPM test was decreased (Figure 2b), again suggesting a basically impaired locomotion. Species-conform behaviors, for example, rearing and digging were also generally reduced in favor of compulsive grooming (Supplementary Table 1 and Supplementary Figure 1). In the LDB test, SPRED2 KO mice again preferred the stressful environment and spent more time in the brightly lit chamber. The latency to cross to the save and dark compartment was prolonged, again indicating a less anxious phenotype in SPRED2 KOs (Figure 2c). Like in the OF test, the number of grooming bouts was higher in SPRED2 KO mice compared with WTs (Supplementary Table 1). We also examined the anxiety behavior in a group of younger male mice aged 2–4 months, which did not show the grooming phenotype or skin lesions. However, we could not detect any behavioral changes of these younger SPRED2 KO mice in the above described tests compared with WT controls (Supplementary Table 2). Therefore, we conclude that reduced anxiety of SPRED2 KO mice correlates with the occurrence of obsessive-compulsive grooming.

Obsessive-compulsive grooming is accompanied by altered anxiety-like behavior and can be alleviated by fluoxetine in SPRED2 KOs. (a) In the open field, the number of grooming bouts was elevated in SPRED2 KO mice (n=9), whereas the total distance traveled by SPRED KOs was reduced compared with WT controls (n=7). (b) In the elevated plus maze, SPRED2 KOs (n=9) spent more time in the open arms and the distance traveled was again reduced in comparison to WT littermates (n=7). (c) In the light/dark box, SPRED2 KO mice (n=9) spent more time in the brightly lit chamber than the WT controls (n=7) and took longer to cross from the lit to the dark compartment. (d) Compared with WTs (n=5), SPRED2 KO mice (n=7) showed neither differences in withdrawal thresholds of the hindpaw upon mechanical stimulation with von Frey filaments nor in hindpaw withdrawal latencies upon thermal stimulation with radiant heat. (e) Photo documentations of mice within the placebo group revealed an unaltered or even worsened state of facial lesions in SPRED2 KO mice (n=7) but no occurence of wounds in WT controls (n=6) after 2 weeks. (f) Photo documentations of mice treated with fluoxetine for 2 weeks demonstrated a clear recovery of occurrence and severity of self-inflicted lesions due to reduced hemorrhages and ulcerations in SPRED2 KOs (n=11) but no visible effects of treatment in WT mice (n=7). (g-h) Videotaping of single mice before and after 2 weeks of fluoxetine treatment revealed an increased duration of grooming events in SPRED2 KO mice (n=11) as compared with WT (n=7) at baseline. (g) Duration of grooming events was not affected by placebo treatment in WT mice but seemed slightly increased in SPRED2 KOs (P=0.073) after 2 weeks. (h) Fluoxetine treatment had no effect on grooming time in WT mice but decreased it in SPRED2 KOs after 2 weeks of treatment. Data are mean±s.e.m; *P<0.05, **P<0.01, ***P<0.001. KO, knockout; n.s., not significant; WT, wildtype.
SPRED2 KO mice did not stop grooming even when they already had apparent lesions. Therefore, we investigated the sensitivity to thermal and mechanical stimuli in SPRED2 KOs and WT controls. We used 7–10 months old mice without apparent lesions and only showing first signs of OCD-like behavior. SPRED2 KOs displayed similar heat and mechanical paw withdrawal latencies and thresholds compared with WT littermates, suggesting intact skin sensitivity (Figure 2d). Since nociceptive testing is normally conducted in younger mice and not well suited for mice permanently engaged in self-grooming, assays were also performed with mice aged 2-3 months. Again, we observed no differences in nociception between young WT and SPRED2 KOs (Supplementary Table 2).
Fluoxetine treatment reduces obsessive-compulsive grooming in SPRED2 KO mice
We next evaluated whether drugs used to treat OCD in humans would be effective in reducing the abnormal grooming in SPRED2 KO mice. Because SSRIs are a first-line treatment for OCD, we treated SPRED2 KO mice with apparent OCD-like phenotype and WT controls aged 7–10 months either with fluoxetine or placebo for 2 weeks. We monitored treatment effects on behavior and the occurrence of skin lesions by photo and video documentations. Photo monitoring revealed no occurrence of lesions in placebo-treated WT mice. In contrast, in placebo-treated SPRED2 KO mice facial lesions resulting from overgrooming remained or ulcerations and hemorrhages even worsened (Figure 2e). In the fluoxetine group, no treatment effects were visible in WT mice, while in SPRED2 KOs the occurrence and severity of self-inflicted lesions were diminished (Figure 2f).
These findings were confirmed by video recordings of single mice at baseline and after 2 weeks of placebo vs fluoxetine treatment. Here, ANOVA revealed a significant time × treatment × genotype interaction (P=0.008) and a highly significant main effect of genotype (P<0.001) for the time mice were engaged in grooming. Overall, SPRED2 KO mice did not only display a higher number of grooming events (Figure 2a) but also a longer grooming time compared with WT mice. Neither placebo nor fluoxetine treatment affected the duration of grooming events in WT mice (Figure 2g and h). In placebo-treated KO mice, however, grooming time seemed slightly increased after 2 weeks compared with baseline (Figure 2g), which is in line with aggravation of facial lesions over time. Interestingly, fluoxetine-treated SPRED2 KO mice spent less time grooming after 2 weeks of treatment compared with baseline (Figure 2h). This confirms that grooming can be interpreted as an OCD-like behavior in SPRED2 KOs, which can be treated with fluoxetine.
Digging is a species-typical behavior in mice but responsive to SSRI treatment.44 Therefore, it can also be regarded as OCD-like and is commonly used as control parameter for fluoxetine effects. Digging was reduced by fluoxetine treatment in addition to excessive grooming, confirming the efficiency of fluoxetine treatment and the OCD-like nature of the self-grooming behavior in SPRED2 KOs (Supplementary Figure 1).
Changed synaptic transmission at cortico-striatal and thalamo-amygdala synapses in SPRED2 KO mice
The pathogenesis of OCD is associated with dysregulation in CSTC circuits. SPRED2 is highly expressed in cortex, striatum, and thalamus but also in other parts of the central nervous system. To detect possible defects in striatal neurotransmission caused by SPRED2 deficiency, we performed whole cell patch clamp measurements on acute brain slices of SPRED2 KO mice with apparent OCD phenotype and WT controls aged 6–8 months. The stimulation electrode was placed in fibers of the corpus callosum and the recording electrode in single neurons of the putamen, which is part of the striatum (Figure 3a). We stimulated putamen-innervating corpus callosum fibers with two consecutive pulses of 1 mA and with an interstimulus interval of 50 ms. This presynaptic fiber stimulation elicited two elevated excitatory postsynaptic currents (EPSCs) in the putamen of SPRED2 KO mice, indicating an increased synaptic transmission at cortico-striatal synapses compared with WT controls (Figure 3b and d). According to that, the stimulation threshold tended to be reduced in putamen neurons of SPRED2 KOs (Figure 3c). Since both EPSC1 and EPSC2 were elevated to a similar extent in SPRED2 KO mice (Figure 3d), the paired pulse ratio in KOs was comparable to WTs (Figure 3e), indicating no relevant changes in the presynaptic vesicle release probability.
The effects seen in cortico-striatal neurotransmission, however, were even more pronounced in thalamo-amygdala circuits. Accordingly, we performed another set of whole cell patch clamp measurements by placing the stimulation electrode in thalamic fibers and the recording electrode in single neurons of the lateral amygdala (Figure 3f). Stimulation of thalamic afferents with two consecutive pulses of 1 mA and with an interstimulus interval of 50 ms elicited an elevated EPSC1 in lateral amygdala neurons of SPRED2 KO mice (Figure 3g and i). This indicates an increased transmission also at thalamo-amygdala synapses and is in line with the reduced stimulation threshold in lateral amygdala neurons of SPRED2 KOs (Figure 3h). The provoked EPSC2 was comparable in WTs and SPRED2 KOs (Figure 3g and i). This leads to a reduced paired pulse ratio in SPRED2 KO mice and reflects changes in the presynaptic vesicle release probability at these amygdaloid synapses (Figure 3j). Therefore, we additionally measured response parameters of spontaneously released vesicles by recording mEPSCs (Figure 3k) in lateral amygdala neurons. The frequency of mEPSCs recorded within 1 min was reduced in SPRED2 KO mice, which again indicates presynaptic alterations in vesicle release probability (Figure 3l). mEPSC magnitudes were also decreased in SPRED2 KOs, which might either be caused by changes in postsynaptic sensitivity or in vesicle transmitter load (Figure 3m).
Altered morphology in lateral amygdala neurons is accompanied by dysregulated transcription and expression of synaptic genes
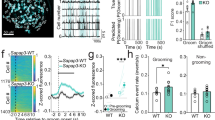
Changes in synaptic input are correlated with morphological alterations in the respective neurons. Hence, we reconstructed pyramidal neurons from the lateral amygdala of SPRED2 KO mice showing apparent OCD-like behavior and of WT controls aged 9–12 months. We detected a higher total spine number on dendritic branches of branch orders 1–4 in SPRED2 KOs (Figure 4a). Spine density per 10 μm of dendritic length was also elevated across these branch orders compared with WTs (Figure 4b).

Differences in lateral amygdala neuron morphology are associated with dysregulated gene transcription and protein expression. (a) Morphological analysis of reconstructed neurons from lateral amygdala of WT (n=26) and SPRED2 KO mice (n=22) revealed a higher total spine number on dendritic branches of branch orders 1–4. (b) Spine density per 10 μm of dendritic length was also higher within these branch orders. (c) Exemplary Western blot analyses showed changed expression levels of different pre- and postsynaptic proteins in the amygdala of SPRED2 KO mice in comparison to WT controls. Protein levels of PSD95, mGluR5, mGluR2, and ERC1 were increased, those of bassoon were decreased. Protein expression of Rab3A, Rab6, α-tubulin, β-tubulin and GAPDH was unaltered; GAPDH was used as loading control. (d) Quantified signals of investigated synaptic proteins after normalization to GAPDH confirmed changed expression levels in the amygdala of SPRED2 KOs (n=11) compared with WTs (n=11). (e) Quantitative RT-PCRs with mRNA isolated from amygdala of SPRED2 KO mice (n=11) and WT controls (n=11) using gene-specific primers confirmed the increased expression of PSD95, mGluR5, mGluR2, and ERC1 on mRNA level. Data are mean±s.e.m; *P<0.05, **P<0.01, ***P<0.001. KO, knockout; n.s., not significant; WT, wildtype.
Since SPREDs are suppressors of Ras/ERK-MAPK signaling and thus critical regulators of cell proliferation and gene expression, we further examined whether changed synaptic excitability and neuron morphology was associated with altered expression of synaptic proteins in the amygdala. Western blot analyses of pre- and postsynaptic proteins demonstrated different expression levels in 10–12 months old SPRED2 KO mice compared with WT controls (Figure 4c). The expression of PSD95, an important anchor of various synaptic proteins at the postsynapse, was upregulated in SPRED2 KOs compared with WT littermates. Protein levels of metabotropic glutamate receptor 5 (mGluR5), which is primarily located at the periphery of postsynaptic densities, were also elevated in SPRED2 KO mice as well as levels of mGluR2, which is primarily distributed at presynaptic axon terminals but may also be expressed at postsynaptic sites. ERC1 (ELKS/RAB6-interacting/CAST family member 1), a structural and functional determinant of the presynaptic active zone, was also upregulated, while presynaptic bassoon, a direct interaction partner of ERC1 in the active zone, was downregulated. The expression of the small GTPases Rab3A and Rab6, which are involved in the regulation of synaptic vesicle transport along microtubules and exocytosis at the presynapse, was not altered. Furthermore, levels of α- and β-tubulin were unchanged (Figure 4c). Quantification of protein amounts by normalization to GAPDH confirmed the dysregulated expression of various pre- and postsynaptic proteins in the amygdala of SPRED2 KOs (Figure 4d).
Differences in protein expression are mainly caused either by altered gene transcription or by protein turnover rate. To address a possible dysregulation at transcriptional level, we performed quantitative RT-PCRs using RNA from the amygdala and gene-specific primers for PSD95, mGluR2, mGluR5 and ERC1. We detected that the dysregulated protein expression observed in Western blots was accompanied by altered expression levels of the selected genes (Figure 4e). Because mRNA and protein levels were changed in the same direction, transcriptional dysregulation is most likely causative for alterations in synaptic protein expression.
SPRED2 deficiency leads to increased TrkB/ERK-MAPK signaling and induces OCD-like grooming in SPRED2 KO mice
Given the dysregulated synaptic gene expression in the amygdala, we focused on the upstream regulatory mechanism that might contribute to the molecular and physiological changes at amygdaloid synapses. An essential regulator of neuronal gene transcription, proliferation and differentiation but also of synaptic transmission and potentiation is the BDNF/TrkB signaling pathway. Upon binding of the neurotrophin BDNF to its preferred receptor tyrosine kinase TrkB, downstream signals are mediated by the Ras/ERK-MAPK cascade. Because SPRED2 is a critical inhibitor of Ras/ERK-MAPK signaling, we investigated if SPRED2 deficiency impacts BDNF/TrkB/ERK-MAPK signaling in SPRED2 KO mice. We analyzed the expression and phosphorylation of ERK1/2 in amygdala of 10–12 months old SPRED2 KO mice and WT littermates by Western blot. The expression of unphosphorylated ERK was not altered, however, P-ERK was increased in SPRED2 KO mice. The 2.5-fold elevated P-ERK/ERK ratio clearly reflected pathway overactivation due to the loss of SPRED2-mediated inhibition (Figure 5a). To unravel whether the increase in Ras/ERK-MAPK signaling is also involved in the development of the OCD-like behavior in SPRED2 KOs, we specifically blocked the Ras/ERK-MAPK in vivo using the MEK1/2 inhibitor selumetinib. In this prospective study, we treated SPRED2 KO mice aged 8 months with selumetinib for one week. Photo documentation revealed reduced hemorrhage and ulceration of self-inflicted wounds in SPRED2 KOs after one week of treatment (Figure 5b). This indicated that SPRED2, as an endogenous suppressor of Ras/ERK-MAPK signaling, is required to ensure normal behavior. Investigation of factors further upstream in this pathway revealed that also Ras was activated 1.9-fold in the amygdala of SPRED2 KO mice compared with WTs (Figure 5c). To test whether pathway activation is elicited by elevated BDNF expression, we estimated BDNF levels in the amygdala of 6–12 months old SPRED2 KO mice and WT controls and found no differences between genotypes (Figure 5d). SPRED2 can be phosphorylated at various confirmed tyrosine residues and might therefore be a direct target of TrkB. To address this, we immunoprecipitated tyrosine-phosphorylated proteins from BDNF-stimulated murine hypothalamic cells and analyzed SPRED2 and TrkB by Western blot. The input controls demonstrated constant expression of both TrkB and SPRED2 in mHypoE44 cells after different times of BDNF stimulation. In the IP samples, BDNF stimulation resulted in increasing TrkB phosphorylation over time. After 60 min of BDNF stimulation, BDNF-mediated TrkB activation provoked phosphorylation of SPRED2 (Figure 5e). Consequently, SPRED2 is a target of TrkB itself or of a kinase downstream of TrkB. Given the unaltered BDNF levels in the amygdala and the interaction between SPRED2 and TrkB, we further investigated whether the augmented activity of Ras and ERK in SPRED2 KO mice might be a result of specifically increased TrkB activation. We used a phospho-RTK array to identify active phosphorylated RTKs in amygdala of 10 months old SPRED2 KO and WT mice. Independently of the genotype, only PDGF-Rα was markedly phosphorylated among the 39 different murine RTKs included in the array. In the amygdala lysates of SPRED2 KOs, we detected highest phosphorylation levels in EGFR, ErbB2 and TrkB (Figure 5f). This demonstrated activation of the TrkB receptor in response to loss of SPRED2; however, a parallel phosphorylation of EGFR and ErbB2 might contribute to induction of downstream pathways. To support our hypothesis that TrkB is responsible for activation of the Ras/ERK-MAPK pathway, we examined possible alterations in TrkB phosphorylation and expression quantitatively. TrkB expression levels were 1.4-fold higher in SPRED2 KO amygdala after normalization to GAPDH in comparison to WT controls (Figure 5g). In addition to TrkB receptor overexpression, phosphorylation of Y515, the tyrosine residue indicative for Ras/ERK-MAPK pathway activation in mouse TrkB, was 1.3-fold elevated in amygdala of SPRED2 KO mice. In contrast, Y817 phosphorylation level, which is relevant for phospholipase C activation, was not altered, indicating that the downstream actions of activated TrkB are specifically mediated by the Ras/ERK MAPK pathway (Figure 5g). Although a contribution of activated EGFR and ErbB2 cannot be excluded, TrkB seems to be a crucial modulator of upregulated ERK-MAPK pathway in SPRED2 KO mice as demonstrated by TrkB overexpression, activation, and association with phosphorylation of SPRED2. Missing inhibition of BDNF/TrkB/ERK-MAPK signaling resulted in OCD-like behavior in SPRED2 KO mice whereas SPRED2-mediated pathway downregulation seems to be necessary for coordinated neuronal protein expression, synaptic function and behavior in vivo.

Increased TrkB/ERK-MAPK signaling caused by SPRED2 deficiency leads to OCD-like grooming in SPRED2 KO mice. (a) Western blot analyses of ERK expression and phosphorylation in amygdala of SPRED2 KO mice (n=8) and WT littermates (n=8) revealed no differences in levels of unphosphorylated ERK but increased levels of phosphorylated ERK in SPRED2 KOs. Upregulation of ERK-MAPK signaling was verified by the 2.5-fold elevated P-ERK/ERK ratio. (b) In vivo inhibition of Ras/ERK-MAPK signaling in SPRED2 KO mice (n=3) by administration of the MEK1/2 inhibitor selumetinib for one week alleviated hemorrhage and ulceration of facial lesions. (c) Ras activation was 1.9-fold higher in SPRED2 KO amygdala (n=8) compared with WT (n=8). (d) BDNF levels in relation to total protein content were unchanged in amygdala lysates of WT (n=18) and SPRED2 KO mice (n=24). (e) Western blot analyses (IB) of SPRED2 and TrkB after BDNF stimulation of murine hypothalamic cells revealed constant TrkB and SPRED2 expression over time (Input). After immunoprecipitation of tyrosine-phosphorylated (PY) proteins from BDNF-stimulated mHypoE44 cells, Western blot analyses detected increasing phosphorylation of TrkB over time and, mediated by active TrkB, phosphorylation of SPRED2 60 min after BDNF stimulation (IP). (f) Parallel determination of phosphorylation of 39 different mouse receptor tyrosine kinases revealed a higher phosphorylation of TrkB specifically in amygdala of SPRED2 KO mice (n=2 for WT and KO). (g) Western blot analyses of TrkB in amygdala indicated an increased expression in SPRED2 KOs reflected by the 1.4-fold TrkB/GAPDH ratio as compared with WTs. Phosphorylation of TrkB at Y515 was 1.3-fold elevated but unchanged at Y817. n=8 for WT and KO. Data are mean±s.e.m; **P<0.01, ***P<0.001. BDNF, brain-derived neurotrophic factor; KO, knockout; n.s., not significant; OCD, obsessive-compulsive disorder; TrkB, tropomyosin receptro kinase B; WT, wildtype.
Discussion
Identification of SPRED2 as a new factor for the pathogenesis of OCD
Our study demonstrates that deficiency of SPRED2 causes excessive and pathological self-grooming in mice. One first clear indication for the presence of an OCD in SPRED2 KOs is that they do not stop grooming even if they already have severe facial lesions with ulcerations and hemorrhage. Reactions to mechanical and heat stimuli were not altered in SPRED2 KO mice, suggesting normal skin sensitivity but compulsive actions up to the point of being self-injurious. Increased anxiety is a common feature associated with certain forms of OCD and also occurs in SAPAP3 or SLITRK5 KO mice, two comparable models of OCD-like grooming.45, 46 Unexpectedly, SPRED2 KO mice did not show increased anxiety-like behavior but seemed less anxious. However, in contrast to the other mouse models, behavioral tests revealed a generally decreased locomotor activity in SPRED2 KOs in addition to the reduced anxiety, which may also be interpreted as lack of drive, lethargy, or signs of depression-like behavior. In line with this, other species-conform behaviors, for example, rearing, were also reduced, indicating that SPRED2 KO mice are massively captured by compulsive actions, which influence experimental readouts. Furthermore, SPRED2 KO mice might also display learning defects similar to mice deficient for the related SPRED1 (ref. 31) and SPRED1-deficient humans.30 Altogether, these disabilities possibly impact appropriate reactions to anxiogenic stimuli or their interpretation. OCD in humans is to a great extent associated with anxiety, specifically avoidance. However, common comorbid conditions also include other psychiatric disorders. Estimated comorbidity with generalized anxiety disorder ranges from below 20% to nearly 76% (overview in47, 48). To a similar extend, OCD is associated with major depressive disorder (40–65% according to47, 48). This may also be the case in SPRED2 deficient mice and explain the reduced locomotor activity and the observed lack of drive. Moreover, OCD and anxiety disorders vary greatly in features like neurocircuitry, neurochemistry, symptoms and cognitive-emotional processing,49 and therefore in DSM-V (5th edition of the Diagnostic and Statistical Manual of Mental Disorders), OCD is no longer categorized as an anxiety disorder, thus emphasizing the differences between both entities.50
A second important feature of OCD is its susceptibility to stress, that is, symptoms increase at times of stress and stressful events may precede the onset of OCD.51, 52 Accordingly, increased stress hormone levels such as corticotropin-releasing hormone, adrenocorticotropic hormone, and corticosterone are often detected in OCD patients.53, 54 This is in line with our previous findings of a hyperactive HPA axis in SPRED2 KO mice, leading to elevated release of these stress hormones.33
Third, SPRED2 KOs were responsive to fluoxetine, a treatment shown to be effective in the reduction of OCD-like grooming and digging.7, 44, 45, 46 Altogether, these data identify SPRED2 as a candidate factor involved in the pathogenesis of OCD-like disorders and SPRED2 KO mice as a suitable new model to investigate OCD-like behaviors.
Excitability changes at cortico-striatal and thalamo-amygdala synapses contribute to OCD pathogenesis in SPRED2 KO mice
Both neuroimaging studies in humans5, 6 and studies with mutant mice implicated dysregulation within CSTC circuits in the pathogenesis of OCD. Mice deficient for SAPAP3 or SLITRK5, like SPRED2 highly expressed in the striatum, displayed alterations in the activity of cortico-striatal neurons in combination with OCD-like grooming.45, 46 In SPRED2 KO mice, we detected increased transmission at cortico-striatal synapses. In line with our results, repeated experimental cortico-striatal stimulation in mice also provoked excessive grooming,7 indicating that especially hyperactivity of cortico-striatal synapses might be causative for OCD-like behaviors and that SPRED2 is crucially involved in regulation of cortico-striatal circuit activity.
Interestingly, we also detected elevated synaptic excitability at thalamo-amygdala synapses of SPRED2 KO mice. Although aberrant function of CSTC circuits is at present the most widely accepted neurobiological explanation for OCD, the underlying pathology is not necessarily limited to orbitofronto-striatal regions. Recent evidence suggests that limbic structures such as hippocampus, anterior cingulate, and amygdala contribute to the pathology of OCD.10, 55, 56 In fact, functional magnetic resonance imaging studies in humans correlate OCD disorders with elevated amygdala activity,55, 56, 57 which is in line with the observed hyperactivity in the lateral amygdala of SPRED2 KO mice. Especially the basolateral amygdala sends prominent projections to the ventral striatum,8, 9, 56 underlining the functional connectivity of both brain regions. Since the amygdala is central to processing emotion and to multiple aspects of cognition that are impaired in OCD, aberrant communication between amygdala and striatum could mediate compulsive behavior. Additionally, dysregulation of amygdala activity may contribute to compulsivity by imparting excessive affective influence on behavioral selection. Here, we provide a mouse model showing similar changes of synaptic excitability in both functionally related brain regions associated with OCD, which is unique amongst previously published mouse models.45, 46, 58 These data suggest that the altered activity of CSTC circuits observed in OCD may indeed be triggered upstream by changes in amygdala activity and that SPRED2 is a crucial modulator of synaptic transmission from thalamus to lateral amygdala.
Changed synaptic input into lateral amygdala results from a combination of altered neuron morphology and dysregulated expression of pre- and postsynaptic proteins
Detailed analysis of electrophysiological data revealed both pre- and postsynaptic changes in SPRED2 KO mice, which contributed to the observed increased synaptic transmission in amygdala and striatum. In the lateral amygdala, diminished paired pulse ratio and decreased frequency of spontaneously released vesicles indicate a reduced presynaptic vesicle release probability. In line with these findings we detected altered expression of ERC1 and bassoon, both scaffolding proteins interacting at the active zone of presynapses. They are not only critical for the integrity of active zone structures,59 but also for the regulation of presynaptic neurotransmitter release.60, 61 Especially ERC1, a homolog of Drosophila's bruchpilot, is required to maintain Ca2+-channel density and synaptic transmission after evoked stimuli,60 which is in line with the ERC1 overexpression and increased excitability of amygdala synapses in SPRED KOs. Consistent with the changes in synaptic transmission, we also found dysregulated expression of glutamate receptors in the amygdala. Because SPRED2 is rather associated with intracellular signaling than with regulation of ion channels, we focused on metabotropic glutamate receptors and revealed a higher expression of mGluR5 and mGluR2. While mGluR2 can be located at both pre- and postsynaptic sites and generally decreases neuronal excitability, mGluR5 acts exclusively at postsynaptic sites by increasing neuronal excitability.62 Dysregulation of mGluRs is generally associated with different psychiatric and anxiety-related disorders, and particularly mGluR5 antagonists and mGluR2 agonists are promising antipsychotic compounds.63 Hence, the observed higher expression of mGluR5 in SPRED2 KOs might contribute to the OCD-like behavior and increased synaptic transmission, whereas the upregulated mGluR2 expression could already be a counterregulatory mechanism and associated with the reduced anxiety. PSD95 is the major scaffolding protein at the postsynapse of mature glutamate synapses and clusters with ionotropic but also with metabotropic glutamate receptors.64 This coupling explains the higher expression of both mGluRs and PSD95 at amygdala postsynapses. Alterations in the expression of PSD95 or of interacting proteins are again associated with a variety of mood disorders in humans,65 in mice especially with OCD-like behaviors, as also observed in the SAPAP3 KO mouse model.45
Taken together, the mostly presynaptic effects detected by electrophysiological recordings in the amygdala are correlated with the dysregulated expression of proteins that have been shown to regulate presynaptic active zone structure and neurotransmitter release like bassoon, ERC1 and mGluR2. The dysregulation of proteins expressed at the postsynapse, for example, PSD95 and mGluR5, is consistent with the detected higher branch order-dependent spine number and spine density in lateral amygdala neurons of SPRED2 KO mice. In fact, PSD95, mGluR5 and also TrkB/ERK-MAPK signaling are associated with regulation of spine morphology, primarily investigated in hippocampus66, 67, 68, 69 but also in amygdala.70
Dendritic spines are the main target for excitatory inputs. Excitatory drive from thalamus to lateral amygdala was increased in SPRED2 KOs as confirmed by higher provoked EPSCs and reduced stimulation threshold. Therefore, higher spine number and density in SPRED2 KOs is supposed to be the structural manifestation of enhanced excitability at thalamo-amygdala synapses. This in turn might contribute to general amygdala hyperactivity, which is a feature of OCD but also of other anxiety disorders.55, 56, 57 Although SPRED2 KO mice did not show increased anxiety in the conducted standard tests, other features of anxiety-related behavior are highly apparent, including increased stress hormone levels due to HPA axis hyperactivity,33 and reduced exploratory locomotion. Accordingly, the amygdala has a high density of CRH and glucocorticoid receptors and stress exposure can induce both amygdala activation and increases in spine density.9, 42, 71 Taken together, the altered synaptic excitability in the amygdala contributes to the OCD-like phenotype in SPRED2 KO mice and is a result of both pre- and postsynaptic effects. Changes in thalamo-amygdala synaptic transmission are caused by dysregulated protein expression as a consequence of altered gene transcription, altogether a result of SPRED2 deficiency and the associated upregulation of TrkB/ERK signaling.
Increased TrkB/BDNF-ERK pathway activity is associated with OCD-like behavior in SPRED2 KO mice
In combination with the OCD-like behavior, we detected hyperactivation of TrKB/ERK-MAPK signaling at critical levels of the cascade. In amygdala of SPRED2 KO mice, TrkB was not only overexpressed, but also phosphorylation of TrkB was increased, as demonstrated by a phospho-RTK-array and by Western blots. More precisely, phosphorylation at Y515 was elevated in amygdala lysates of SPRED2 KO mice, phosphorylation at Y817, however, was unchanged. Whereas P-TrkB Y817 is responsible for induction of downstream phospholipase C-mediated cascades, P-TrkB Y515 triggers Ras/ERK-MAPK pathways,13 indicating that they are specifically activated by active TrKB in SPRED2 KOs. Consequently, both Ras activity and ERK phosphorylation were augmented in SPRED2 KO amygdala. BDNF levels in amygdala were not changed, demonstrating that overactivation of Ras/ERK-MAPK signaling in fact results from loss of SPRED2-mediated pathway inhibition.
Furthermore, we detected that SPRED2 is phosphorylated after BDNF-stimulated TrkB activation. TrkB is activated by autophosphorylation and in turn activates various downstream targets by phosphorylation. SPRED2 can be phosphorylated at different tyrosine residues in response to stimulation by various growth factors.23 SPRED phosphorylation is functionally required for regulation of the inhibitory effect72, 73 or as prevention of proteasomal degradation.72 We assume that TrkB-dependent tyrosine phosphorylation of SPRED2 might be preventive for protein degradation and might enable the suppression of Ras/ERK signaling stimulated by BDNF-induced TrkB activation. This mechanism could regulate SPRED2 steady-state levels, maintain physiological TrkB/BDNF-ERK pathway activity in vivo, and ensure normal behavior.
BDNF/TrkB pathways are crucially involved in nearly all stages of neural circuit development and associated with multiple neuropsychiatrc diseases.12, 13, 14, 15 Untill now, however, only first descriptive studies associate the BDNF/TrkB system with OCD16, 17 or related disorders in humans and mice, indicating that genetic mutations and altered protein levels might play a role.18, 19, 20 ERK, a critical regulator of proliferation, is present in presumably all cells and tissues. In brain, ERK contributes to the induction of transcription of plasticity-related genes, mediates synaptic transmitter release and is therefore implicated in learning and memory.74, 75, 76 However, not much is known about the role of ERK in neuropsychiatric disorders, especially in OCD-like disorders. In SPRED2 KO mice, we determined the mechanism how SPRED2 deficiency leads to an increase of active TrkB receptor and signaling, which is mediated downstream by increased Ras activity and ERK-phosporylation and induces OCD-like behavior.
The contribution of dysregulated TrkB/ERK-MAPK signaling to OCD-development was confirmed by administration of selumetinib, a MEK1/2 inhibitor and specific blocker of Ras/ERK-MAPK activity. Selumetinib is experimentally used for the treatment of cancer in mouse models and clinical trials have also been conducted in humans.77, 78 Although we used selumetinib not as a putative medication but as an experimental rescue, it robustly alleviated self-inflicted lesions resulting from excessive grooming in SPRED2 KO mice. Our prospective in vivo study showed that the TrkB-activated ERK-MAPK pathway is specifically involved in the development of OCD and confirmed that physiological SPRED2-regulated pathway activity is required to maintain normal behavior in vivo.
Here we provide evidence that upregulation of the Ras/ERK-MAPK pathway is not only involved in cancer pathogenesis and developmental disorders like rasopathies, but also in the development of OCD-related disorders. We ascertained thalamo-amygdala circuits as affected brain region in addition to the known cortico-striatal circuitry. In the amygdala, the upstream trigger of OCD-like behavior is hyperactivity of BDNF/TrkB signaling, a result of the loss of SPRED2-mediated pathway inhibition. BDNF/TrkB/ERK-MAPK pathway dysregulation leads to changes in pre- and postsynaptic mRNA and protein expression and to alterations of thalamo-amygdala synaptic transmission. Both the SSRI fluoxetine and the Ras/ERK pathway inhibitor selumetinib reduced OCD-like grooming in SPRED2 KO mice (Supplementary Figure 2). Thus, our study identifies SPRED2 as a considerable factor in the pathogenesis of OCD, as a critical regulator of synaptic transmission in different brain regions and as a new regulator of BDNF/TrkB pathways. SPRED2 is highly conserved, the most ubiquitously expressed SPRED family member, and its expression is especially widespread in brain. Therefore, SPRED2 is a very promising target for further and more specific studies of brain function and associated neuropsychiatric, -developmental and -degenerative diseases both in mice and humans.
References
Leckman JF, Grice DE, Boardman J, Zhang H, Vitale A, Bondi C et al. Symptoms of obsessive-compulsive disorder. Am J Psychiatry 1997; 154: 911–917.
Miguel EC, Leckman JF, Rauch S, do Rosario-Campos MC, Hounie AG, Mercadante MT et al. Obsessive-compulsive disorder phenotypes: implications for genetic studies. Mol Psychiatry 2005; 10: 258–275.
Fineberg NA, Potenza MN, Chamberlain SR, Berlin HA, Menzies L, Bechara A et al. Probing compulsive and impulsive behaviors, from animal models to endophenotypes: a narrative review. Neuropsychopharmacology 2010; 35: 591–604.
Browne HA, Gair SL, Scharf JM, Grice DE . Genetics of obsessive-compulsive disorder and related disorders. Psychiatr Clin North Am 2014; 37: 319–335.
Graybiel AM, Rauch SL . Toward a neurobiology of obsessive-compulsive disorder. Neuron 2000; 28: 343–347.
Aouizerate B, Guehl D, Cuny E, Rougier A, Bioulac B, Tignol J et al. Pathophysiology of obsessive-compulsive disorder: a necessary link between phenomenology, neuropsychology, imagery and physiology. Prog Neurobiol 2004; 72: 195–221.
Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K et al. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science (New York, NY) 2013; 340: 1234–1239.
Janak PH, Tye KM . From circuits to behaviour in the amygdala. Nature 2015; 517: 284–292.
Davis M, Whalen PJ . The amygdala: vigilance and emotion. Mol Psychiatry 2001; 6: 13–34.
Milad MR, Rauch SL . Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cogn Sci 2012; 16: 43–51.
Taylor S . Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol Psychiatry 2013; 18: 799–805.
Huang EJ, Reichardt LF . Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001; 24: 677–736.
Huang EJ, Reichardt LF . Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 2003; 72: 609–642.
Autry AE, Monteggia LM . Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 2012; 64: 238–258.
Zuccato C, Cattaneo E . Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 2009; 5: 311–322.
Hall D, Dhilla A, Charalambous A, Gogos JA, Karayiorgou M . Sequence variants of the brain-derived neurotrophic factor (BDNF) gene are strongly associated with obsessive-compulsive disorder. Am J Hum Genet 2003; 73: 370–376.
Hemmings SM, Kinnear CJ, Van der Merwe L, Lochner C, Corfield VA, Moolman-Smook JC et al. Investigating the role of the brain-derived neurotrophic factor (BDNF) val66met variant in obsessive-compulsive disorder (OCD). World J Biol Psychiatry 2008; 9: 126–134.
Oliveira-Maia AJ, Castro-Rodrigues P . Brain-derived neurotrophic factor: a biomarker for obsessive-compulsive disorder? Front Neurosci 2015; 9: 134.
Alonso P, Gratacos M, Menchon JM, Saiz-Ruiz J, Segalas C, Baca-Garcia E et al. Extensive genotyping of the BDNF and NTRK2 genes define protective haplotypes against obsessive-compulsive disorder. Biol Psychiatry 2008; 63: 619–628.
Olsen D, Kaas M, Schwartz O, Nykjaer A, Glerup S . Loss of BDNF or its receptors in three mouse models has unpredictable consequences for anxiety and fear acquisition. Learn Mem 2013; 20: 499–504.
Guiton M, Gunn-Moore FJ, Stitt TN, Yancopoulos GD, Tavare JM . Identification of in vivo brain-derived neurotrophic factor-stimulated autophosphorylation sites on the TrkB receptor tyrosine kinase by site-directed mutagenesis. J Biol Chem 1994; 269: 30370–30377.
Zhang W, Liu HT . MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002; 12: 9–18.
Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K et al. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001; 412: 647–651.
King JA, Straffon AF, D'Abaco GM, Poon CL, I ST, Smith CM et al. Distinct requirements for the Sprouty domain for functional activity of Spred proteins. Biochem J 2005; 388 (Pt 2): 445–454.
Bundschu K, Walter U, Schuh K . Getting a first clue about SPRED functions. Bioessays 2007; 29: 897–907.
Bundschu K, Gattenlohner S, Knobeloch KP, Walter U, Schuh K . Tissue-specific Spred-2 promoter activity characterized by a gene trap approach. Gene Expr Patterns 2006; 6: 247–255.
Engelhardt CM, Bundschu K, Messerschmitt M, Renne T, Walter U, Reinhard M et al. Expression and subcellular localization of Spred proteins in mouse and human tissues. Histochem Cell Biol 2004; 122: 527–538.
Phoenix TN, Temple S . Spred1, a negative regulator of Ras-MAPK-ERK, is enriched in CNS germinal zones, dampens NSC proliferation, and maintains ventricular zone structure. Genes Dev 2010; 24: 45–56.
Mardakheh FK, Yekezare M, Machesky LM, Heath JK . Spred2 interaction with the late endosomal protein NBR1 down-regulates fibroblast growth factor receptor signaling. J Cell Biol 2009; 187: 265–277.
Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet 2007; 39: 1120–1126.
Denayer E, Ahmed T, Brems H, Van Woerden G, Borgesius NZ, Callaerts-Vegh Z et al. Spred1 is required for synaptic plasticity and hippocampus-dependent learning. J Neurosci 2008; 28: 14443–14449.
Inoue H, Kato R, Fukuyama S, Nonami A, Taniguchi K, Matsumoto K et al. Spred-1 negatively regulates allergen-induced airway eosinophilia and hyperresponsiveness. J Exp Med 2005; 201: 73–82.
Ullrich M, Bundschu K, Benz PM, Abesser M, Freudinger R, Fischer T et al. Identification of SPRED2 (Sprouty-related Protein with EVH1 Domain 2) as a Negative Regulator of the Hypothalamic-Pituitary-Adrenal Axis. J Biol Chem 2011; 286: 9477–9488.
Bundschu K, Knobeloch KP, Ullrich M, Schinke T, Amling M, Engelhardt CM et al. Gene disruption of Spred-2 causes dwarfism. J Biol Chem 2005; 280: 28572–28580.
Ullrich M, Schuh K . Gene trap: knockout on the fast lane. Methods Mol Biol (Clifton, NJ) 2009; 561: 145–159.
Faul F, Erdfelder E, Lang AG, Buchner A . G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 2007; 39: 175–191.
Post AM, Weyers P, Holzer P, Painsipp E, Pauli P, Wultsch T et al. Gene-environment interaction influences anxiety-like behavior in ethologically based mouse models. Behav Brain Res 2011; 218: 99–105.
Bourin M, Hascoet M . The mouse light/dark box test. Eur J Pharmacol 2003; 463: 55–65.
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL . Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53: 55–63.
Hargreaves K, Dubner R, Brown F, Flores C, Joris J . A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988; 32: 77–88.
Paxinos G, Franklin KBJ . The Mouse Brain in Stereotaxic Coordinates. Academic Press, 1997.
Nietzer SL, Bonn M, Jansen F, Heiming RS, Lewejohann L, Sachser N et al. Serotonin transporter knockout and repeated social defeat stress: impact on neuronal morphology and plasticity in limbic brain areas. Behav Brain Res 2011; 220: 42–54.
Chen Y, Boettger MK, Reif A, Schmitt A, Uceyler N, Sommer C . Nitric oxide synthase modulates CFA-induced thermal hyperalgesia through cytokine regulation in mice. Mol Pain 2010; 6: 13.
Huang GJ, Bannerman D, Flint J . Chronic fluoxetine treatment alters behavior, but not adult hippocampal neurogenesis, in BALB/cJ mice. Mol Psychiatry 2008; 13: 119–121.
Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 2007; 448: 894–900.
Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med 2010; 16: 598–602, 591p following 602.
Bartz JA, Hollander E . Is obsessive-compulsive disorder an anxiety disorder? Prog Neuropsychopharmacol Biol Psychiatry 2006; 30: 338–352.
Ruscio AM, Stein DJ, Chiu WT, Kessler RC . The epidemiology of obsessive-compulsive disorder in the National Comorbidity Survey Replication. Mol Psychiatry 2010; 15: 53–63.
Stein DJ, Fineberg NA, Bienvenu OJ, Denys D, Lochner C, Nestadt G et al. Should OCD be classified as an anxiety disorder in DSM-V? Depress Anxiety 2010; 27: 495–506.
Association AP (ed). Diagnostic and statistical manual of mental disorders. 5th Edition. American Psychiatric Publishing: Arlington, VA, 2013.
Findley DB, Leckman JF, Katsovich L, Lin H, Zhang H, Grantz H et al. Development of the Yale Children's Global Stress Index (YCGSI) and its application in children and adolescents ith Tourette's syndrome and obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry 2003; 42: 450–457.
Toro J, Cervera M, Osejo E, Salamero M . Obsessive-compulsive disorder in childhood and adolescence: a clinical study. J Child Psychol Psychiatry 1992; 33: 1025–1037.
Altemus M, Pigott T, Kalogeras KT, Demitrack M, Dubbert B, Murphy DL et al. Abnormalities in the regulation of vasopressin and corticotropin releasing factor secretion in obsessive-compulsive disorder. Arch Gen Psychiatry 1992; 49: 9–20.
Kluge M, Schussler P, Kunzel HE, Dresler M, Yassouridis A, Steiger A . Increased nocturnal secretion of ACTH and cortisol in obsessive compulsive disorder. J Psychiatr Res 2007; 41: 928–933.
Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET . Integrating evidence from neuroimaging and neuropsychological studies of obsessive-compulsive disorder: the orbitofronto-striatal model revisited. Neurosci Biobehav Rev 2008; 32: 525–549.
Wood J, Ahmari SE . A framework for understanding the emerging role of corticolimbic-ventral striatal networks in OCD-associated repetitive behaviors. Front Syst Neurosci 2015; 9: 171.
Breiter HC, Rauch SL, Kwong KK, Baker JR, Weisskoff RM, Kennedy DN et al. Functional magnetic resonance imaging of symptom provocation in obsessive-compulsive disorder. Arch Gen Psychiatry 1996; 53: 595–606.
Albelda N, Joel D . Current animal models of obsessive compulsive disorder: an update. Neuroscience 2012; 211: 83–106.
Sudhof TC . The presynaptic active zone. Neuron 2012; 75: 11–25.
Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A et al. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science (New York, NY) 2006; 312: 1051–1054.
Davydova D, Marini C, King C, Klueva J, Bischof F, Romorini S et al. Bassoon specifically controls presynaptic P/Q-type Ca(2+) channels via RIM-binding protein. Neuron 2014; 82: 181–194.
Benarroch EE . Metabotropic glutamate receptors: synaptic modulators and therapeutic targets for neurologic disease. Neurology 2008; 70: 964–968.
Niswender CM, Conn PJ . Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 2010; 50: 295–322.
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 1999; 23: 583–592.
Keith D, El-Husseini A . Excitation control: balancing PSD-95 function at the synapse. Front Mol Neurosci 2008; 1: 4.
Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL . Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 2008; 60: 788–802.
Vanderklish PW, Edelman GM . Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci USA 2002; 99: 1639–1644.
Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ et al. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci USA 2006; 103: 1071–1076.
Alonso M, Medina JH, Pozzo-Miller L . ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem 2004; 11: 172–178.
Bennett MR, Lagopoulos J . Stress and trauma: BDNF control of dendritic-spine formation and regression. Prog Neurobiol 2014; 112: 80–99.
Roozendaal B, McEwen BS, Chattarji S . Stress, memory and the amygdala. Nat Rev Neurosci 2009; 10: 423–433.
Meng S, Zhang M, Pan W, Li Z, Anderson DH, Zhang S et al. Tyrosines 303/343/353 within the Sprouty-related domain of Spred2 are essential for its interaction with p85 and inhibitory effect on Ras/ERK activation. Int J Biochem Cell Biol 2012; 44: 748–758.
Quintanar-Audelo M, Yusoff P, Sinniah S, Chandramouli S, Guy GR . Sprouty-related Ena/VASP homology 1-domain-containing protein (SPRED) 1, a SHP2 substrate in the Ras/ERK pathway. J Biol Chem 2011; 286: 23102–23112.
Kelleher RJ 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S . Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 2004; 116: 467–479.
Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008; 135: 549–560.
Giese KP, Mizuno K . The roles of protein kinases in learning and memory. Learn Mem 2013; 20: 540–552.
Bartholomeusz C, Xie X, Pitner MK, Kondo K, Dadbin A, Lee J et al. MEK inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung metastasis in a triple-negative breast cancer xenograft model. Mol Cancer Ther 2015; 14: 2773–2781.
Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. J Am Med Assoc 2014; 311: 2397–2405.
Acknowledgements
We thank Lydia Biko for expert technical help with mechanical and thermal sensitivity assays. We are also very grateful to Marion Winning for her excellent technical expertize in Golgi-Cox staining. We acknowledge Marco Abesser for technical assistance in photography and videotaping and Nadine Ehmann and Martin Pauli for generously providing reagents. The position of Melanie Ullrich was funded by the DFG (SCHU1600/3-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ullrich, M., Weber, M., Post, A. et al. OCD-like behavior is caused by dysfunction of thalamo-amygdala circuits and upregulated TrkB/ERK-MAPK signaling as a result of SPRED2 deficiency. Mol Psychiatry 23, 444–458 (2018). https://doi.org/10.1038/mp.2016.232
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.232
This article is cited by
-
Persistent enhancement of basolateral amygdala-dorsomedial striatum synapses causes compulsive-like behaviors in mice
Nature Communications (2024)
-
From compulsivity to compulsion: the neural basis of compulsive disorders
Nature Reviews Neuroscience (2024)
-
Late development of OCD-like phenotypes in Dlgap1 knockout mice
Psychopharmacology (2024)
-
Training-induced circuit-specific excitatory synaptogenesis in mice is required for effort control
Nature Communications (2023)
-
Landscape and future directions of machine learning applications in closed-loop brain stimulation
npj Digital Medicine (2023)
