
Abstract
The adult form of attention deficit/hyperactivity disorder (aADHD) has a prevalence of up to 5% and is the most severe long-term outcome of this common neurodevelopmental disorder. Family studies in clinical samples suggest an increased familial liability for aADHD compared with childhood ADHD (cADHD), whereas twin studies based on self-rated symptoms in adult population samples show moderate heritability estimates of 30–40%. However, using multiple sources of information, the heritability of clinically diagnosed aADHD and cADHD is very similar. Results of candidate gene as well as genome-wide molecular genetic studies in aADHD samples implicate some of the same genes involved in ADHD in children, although in some cases different alleles and different genes may be responsible for adult versus childhood ADHD. Linkage studies have been successful in identifying loci for aADHD and led to the identification of LPHN3 and CDH13 as novel genes associated with ADHD across the lifespan. In addition, studies of rare genetic variants have identified probable causative mutations for aADHD. Use of endophenotypes based on neuropsychology and neuroimaging, as well as next-generation genome analysis and improved statistical and bioinformatic analysis methods hold the promise of identifying additional genetic variants involved in disease etiology. Large, international collaborations have paved the way for well-powered studies. Progress in identifying aADHD risk genes may provide us with tools for the prediction of disease progression in the clinic and better treatment, and ultimately may help to prevent persistence of ADHD into adulthood.
Similar content being viewed by others
Adult ADHD
Attention-deficit/hyperactivity disorder (ADHD) is a common, childhood onset, chronic neuropsychiatric disorder characterized by developmentally inappropriate inattentiveness, increased impulsivity and hyperactivity, impairing multiple areas of life.1 ADHD has long been considered a disorder of childhood that resolves with maturation. Symptoms of inattention, impulsiveness, restlessness and emotional dysregulation in adults were considered not to reflect ADHD, but to be unspecific problems secondary to other disorders. This idea was challenged when systematic follow-up studies of children documented the persistence of ADHD into adulthood.2 Longitudinal follow-up studies of ADHD children, community surveys and epidemiological studies of population samples estimate the average prevalence of adult ADHD (aADHD) to be between 2.5 and 4.9%.3 This shows that aADHD is one of the most common psychiatric disorders in our society and clinical settings. The notion that the total number of people affected by aADHD is even larger than those suffering from ADHD during childhood and adolescence also shows that the societal consequences of this chronic debilitating condition may have been vastly underestimated in the past.4
Clinical research has shown that the predominant features of aADHD differ from typical ADHD in children (cADHD), with less obvious symptoms of hyperactivity or impulsivity and more inattentive symptoms; importantly, the frequency of psychiatric comorbidity is also increased in aADHD.5 Until recently, aADHD has been diagnosed according to clinical descriptions originally developed for children. The lack of age-appropriate clinical measures has hampered progress in this field, including genetic research. Future versions of the Diagnostic and Statistical Manual of Mental Disorders1 may provide diagnostic measures that are better suited for all relevant age groups.
Heritability, family studies, suitability for genetic studies
Family and twin studies of cADHD demonstrate a high heritability, estimated to be around 70–80% from twin studies.6, 7 Relatively few studies have investigated the genetic and environmental contributions to the developmental course and outcomes in adulthood. Longitudinal twin studies show that the continuity of symptoms from childhood through to adolescence is predominantly due to common genetic influences.8, 9, 10 Although such stable genetic effects are likely to continue beyond the adolescent years, there are only a few studies investigating this.
Genetic research on ADHD started with the finding that hyperactivity tends to aggregate in families.11, 12 Since then, family studies have shown that ADHD shows familial clustering both within and across generations. Increased rates of ADHD among the parents and siblings of ADHD children have been observed.13, 14 In addition, strongly increased risks for ADHD (57%) among the offspring of adults with ADHD have been reported.15 Also, compared with the risk for ADHD among the siblings of children with ADHD (15%), siblings of adults with ADHD were found to have a strongly increased ADHD risk (41%).16 Furthermore, a prospective 4-year follow-up study of male children into mid-adolescence found the prevalence of ADHD to be significantly higher among the parents and siblings of persistent ADHD child probands compared with the relatives of ADHD probands in whom ADHD remitted.17 Taken together, these studies suggest that the risk for ADHD may be greater among the first-degree relatives of probands with ADHD that persists into adolescence and adulthood than that among the relatives of probands with ADHD that remits before adulthood.17, 18
Whether such familial risks reflect genetic or environmental factors can be clarified using adoption and twin studies. Adoption studies found that ADHD is transmitted only to biological relatives, which strongly implicates genetic factors as the main causal influences on familial risk for the disorder.11, 12, 19, 20, 21 These studies showed (for both current and retrospective symptoms in adults) that cADHD in child relatives predicts aADHD (or associated symptoms) in adult relatives. However, both adoption and family studies identify discrepancies related to different sources of ratings, with self-evaluation of ADHD symptoms by adults providing less evidence of familial effects than informants or cognitive performance data.19, 22, 23
Recently, four adult population twin studies using self-ratings of ADHD symptoms have been completed, which all found heritabilities that are far lower than those found in similar studies of parent- or teacher-rated cADHD: 41% for retrospectively reported childhood ADHD symptoms in a sample of 345 US veterans aged 41–58 years old,24 40% for current inattention problems in a Dutch study of 4245 18–30-year olds,8 30% for current ADHD symptoms in a Dutch study of over 12 000 twin pairs with an average age of 31 years25 and 35% for current ADHD in a Swedish sample of more than 15 000 twin pairs aged 20–46 years (Larsson et al., unpublished data). The situation is similar in adolescence, as adolescent twin studies using self-ratings show lower heritability estimates than studies of parent or teacher ratings,26, 27 suggesting that self-ratings may be a poorer measure of the underlying genetic liability to ADHD than informant reports or clinical interviews. Although the estimated heritability in self-rated ADHD symptoms in adult populations is lower than that derived from parent or teacher ratings of cADHD, the pattern of findings is identical. Both types of studies find that there are no gender differences observed in the estimates of heritability, heritability estimates are stable across the age-span (for each type of measurement approach), there are similar estimates of the genetic correlation (the proportion of shared genetic effects) of 60–70% between inattention and hyperactivity-impulsivity, familial effects are all genetic in origin with no shared environmental influences, and no threshold effects are found. This suggests that for both child and adult ADHD the disorder is best perceived as the impairing extreme of a quantitative trait (Larsson et al., unpublished data; ref. 28).
Despite these common features, the relatively low heritability estimates for ADHD symptoms in adults derived from population twin studies need some explanation, because they appear to be at odds with heritability estimates of ADHD symptoms in children, as well as the family studies that show a high familial risk for persistent forms of ADHD.15, 18 Several factors are likely involved. We have already mentioned the consistent finding that self-ratings of ADHD symptoms give lower estimates of heritability compared with informant ratings in twin studies. One source of measurement error (that is, variance of the true diagnostic status that is not predicted by the measurement instrument) is the reliability of the self-rated measures of ADHD symptoms. In one of the heritability studies by Boomsma and co-workers,25 this was estimated to be around 0.66, which is lower than the mean heritability of cADHD across extant studies.29 Psychometric studies also show that, although self-ratings may be useful as a screening tool for aADHD, their correspondence with the full diagnosis is only modest. For example, Kessler et al.30 reported that the sensitivity of self-ratings as a measure of diagnosis was high (98%), whereas the specificity was not (56%). Similar findings were reported by Daigre Blanco et al.31 (87.5% sensitivity and 68.6% specificity). Related to this source of error are potential effects of having two raters in twin studies of self-ratings of ADHD (each twin rates him/herself), whereas informants usually rate both members of a twin pair. Since reliability between two raters will always be less than an individual's reliability with their own ratings, and because a ceiling on heritability is set by the reliability of ratings, heritability estimates will always be lower when two separate raters are involved in evaluating each twin pair compared with only one. Single raters may inflate identical twin pair similarities, potentially leading to an overestimation of heritability in the reported studies on cADHD, whereas the lower reliability of ratings between two raters may lead to lower estimates. Evidence for the later conclusion comes from our recent analysis of same versus different teacher ratings in a study of 5641 12-year-old twins, with heritability estimates of 75% for same teacher and 53% for different teacher ratings of twin pairs (Merwood and Asherson, unpublished data).
Another relevant difference between child and adult samples is the expected range of ADHD symptom scores. It is well known that ADHD symptoms decline through adolescence into adulthood.32 Thus, it is possible that the restricted range of ADHD symptoms in adulthood could influence estimates of heritability. Although some of this symptom decline is likely due to true remission of ADHD, some have argued that the diagnostic criteria for ADHD, which were originally developed for children, are developmentally insensitive and thus become less sensitive to ADHD with age (see above and refs. 4,33). Added to this is the possibility that in cross-sectional studies of adult population twin studies (that do not apply clinical criteria for ADHD), ADHD symptoms may emerge in some individuals owing to adult-onset conditions, such as anxiety, depression and drug use. These ‘phenocopies’ would lead to increased measurement error of the genetic liability for ADHD and lower estimates of heritability.
Differences in the way that participants are ascertained in different study designs may also impact on estimates of familial/genetic influences. The family studies that showed high familial risk for ADHD used case–control methods to ascertain adult patients who were self-referred for (severe) ADHD-like problems. There are notable differences between the clinically referred and population-based samples. The former have a more skewed male-to-female ratio, higher rates of psychiatric comorbidity and lower rates of primarily inattentive ADHD. Moreover, the family and twin studies used differing assessment methodologies. The family studies diagnosed subjects with structured interviews that evaluated childhood onset of impairing symptoms and the presence of impairment in multiple settings as required by Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. In contrast, with the exception of Schultz et al.,34 the twin studies used rating scale measures of ADHD symptoms that do not query for childhood onset and do not systematically assess impairment in multiple settings.
Overall, these considerations suggest that the lower heritability of aADHD compared with cADHD could be due to increased measurement error in the aADHD twin studies. Support for this conclusion comes from a recent Swedish twin study, which found that the heritability of attention problems in 19–20 year olds was estimated at 78% when self-rating and parent-rating data were combined; the heritability for self-ratings alone was 48% (Larsson et al., unpublished data). Analogous to this, cluster A personality disorders show low heritability estimates in analyses based on limited phenotypic information that become much higher when adding more information from interviews.35 On the other hand, it still remains feasible that the heritability of aADHD does indeed decline with increasing age. This might reflect the importance of developmental processes that are sensitive to person-specific environmental factors affecting the longitudinal outcome of ADHD in adults.
Since heritability estimates do not relate directly to the frequency or effect size of specific genetic risk factors,36 it is not yet clear as to what the lower heritability estimates actually mean for molecular genetic studies of aADHD. For example, some disorders with low heritabilities, such as prostate and breast cancer, have identified genes with moderate to large effects,37 yet this is not the case for many highly heritable phenotypes including ADHD.38 In the absence of sufficient studies on this issue, it is quite clear that genetic researchers should preferably use measures that have been shown in family studies to have high rates of familial transmission and in adoption studies to aggregate in biological, rather than adoptive relatives. The evidence for strong familial risks in the relatives of adolescent and adult ADHD probands suggests that the clinical diagnosis of aADHD may represent a more familial measure, although there are no studies to date that directly address this question. The difference could arise because the clinical diagnosis takes a developmental perspective in which the adult phenotype reflects persistence of the childhood disorder, whereas the cross-sectional data used in twin studies may include adult-onset causes of ADHD-like symptoms that reflect phenocopies involving different etiological processes.
We conclude that aADHD is influenced by familial factors that are genetic in origin. The available studies indicate that self-ratings of Diagnostic and Statistical Manual of Mental Disorders, 4th Edition-defined ADHD symptoms may not be the best measure of the underlying genetic risk for aADHD and that other factors such as childhood onset, pervasiveness and impairment should be taken into account.
Molecular genetic studies
Candidate gene association studies in adult ADHD
A search of NCBI's PubMed database for genetic association studies revealed 46 publications on aADHD (published until June 2011). Most of these studies are based on clinically assessed patients. The majority of studies examined single (or a few) polymorphisms in dopaminergic and serotonergic genes focusing predominantly on the dopamine transporter (SLC6A3/DAT1) and the dopamine receptor D4 (DRD4), both associated with cADHD in meta-analysis39 (Table 1).40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83
In all, 10 studies looked at the 40-bp variable number of tandem repeats (VNTR) in the 3′-untranslated region (3′-UTR) of the SLC6A3/DAT1 gene, or a haplotype of this and a second 30-bp VNTR in intron 8. Although most studies found no evidence of association with aADHD,40, 42, 43, 46, 48, 49 three studies41, 44, 47 found a consistent association with the 9-repeat allele or the 9-6 haplotype rather than the 10/10 genotype or the 10-6 haplotype associated with cADHD.84, 85 This association of the 9-6 haplotype and the 9/9 genotype with aADHD was confirmed by a meta-analysis of 1440 cases and 1769 controls,45 which makes it the most robust finding for adult ADHD, to date. Why the association in adults is different from the one found in children is not entirely clear. A number of explanation are possible, for example, (a) that the 9-repeat allele and the 9-6 haplotype may mark a severe subgroup of ADHD patients prone to disease persistence, (b) SLC6A3 may modulate rather than cause ADHD, and (c) that the 9-repeat allele and 9-6 haplotype only become aberrant in an adult brain with its lower dopamine levels.45 For DRD4, most studies predominantly focused on a functional tandem repeat polymorphism in exon 3 of DRD4, in which one variant (the 7-repeat allele) is associated with cADHD.39 Of six studies from five independent samples, three were negative,41, 43, 49 whereas the three others showed nominal evidence that the 7-repeat allele increased risk for aADHD.51, 52, 53 A recent study of the long-term outcome of cADHD suggested that carriers of the 7-repeat allele show a more persistent outcome of ADHD.48 While being reasonably powered, this latter result seems at odds with an earlier finding showing a normalization of the cortical thickness in ADHD-relevant brain regions linked to a better clinical outcome during adolescence in carriers of the DRD4 7-repeat allele.86 A large meta-analysis in 1608 aADHD patient and 2358 control samples was negative for the 7-repeat allele, although showing nominal evidence for association of a haplotype formed by the common 4-repeat allele of the exon 3 VNTR and the long (L) allele of the 120-bp insertion/deletion upstream of DRD4.50 Findings for DRD4 are the most consistent ones for cADHD,39 but more research is clearly needed to understand its role in aADHD. Among studies on other dopamine receptor genes (DRD2, DRD3 and DRD5), the findings for a DRD5 VNTR have been most positive. Although individually unconvincing, the findings of two of three studies point in the same direction, indicating that the same allele associated with cADHD might also increase risk for aADHD.43, 58 Of the genes involved in dopamine turnover, COMT and DBH, the two largest studies (investigating functional COMT variants) showed association with aADHD. However, the direction of association in each of the studies was opposite.63, 69
Among the serotonergic genes, the serotonin transporter gene (SLC6A4/5-HTT/SERT) and its functional polymorphism, 5-HTTLPR, were studied most often,48, 56, 57, 61, 64, 65, 66, 67, 87 with essentially negative or conflicting results, even in a large meta-analysis of 1894 patients and 1977 controls.70 One study used a tagging approach and investigated a total of 19 serotonergic genes68 and reported association of aADHD with single markers or haplotypes in MAOB, DDC and HTR2A. The latter gene was also found associated with aADHD in one of two additional studies, although the polymorphism involved was different.49, 69 A recent, large study in 1636 patients and 1923 controls investigated the two TPH genes and found nominal evidence of association with TPH1, but not TPH2.70
Three genes in the noradrenergic system, the noradrenalin transporter (SLC6A2/NET), ADRA2A and ADRA2C, have been tested for association with aADHD. As shown in Table 1, however, there has been no evidence of association for these genes with aADHD.61, 62, 71, 72, 88
Two studies have looked at several genes encoding neurotrophic factors: Ribasés et al.89 used a full tagging approach and showed association with aADHD for CNTFR, but did not replicate earlier research, suggesting an association with NTF3.74 There have been five studies of the BDNF functional Val66Met polymorphism,49, 74, 76, 90 including a meta-analysis of 1445 cases and 2247 controls,75 that found no evidence for association.
A number of novel candidate genes have been associated with aADHD in isolated studies and are in need of independent replication. Of particular interest may be LPHN3, which was selected for study from fine mapping of a significant linkage region on chromosome 4q13.81 LPHN3 was associated with ADHD in a large sample of children and adults,81 and subsequently replicated in an independent aADHD sample.82 The function of this gene, which encodes a G-protein-coupled receptor, is still not well understood.91 BAIAP2 was found to be associated with aADHD in a study investigating six genes with lateralized expression in the developing brain;73 as brain asymmetry is altered in ADHD,92 this interesting finding might point to etiological pathways for ADHD not involving neurotransmitter signaling. In addition, following up the findings for the circadian rhythm gene CLOCK may be fruitful,78 as a dysregulated circadian rhythm is a consistent finding in both cADHD and aADHD.93 A fourth gene, NOS1, associated with aADHD80 should be considered in future studies, as evidence for an involvement of this gene in ADHD also comes from a genome-wide association study (GWAS) in cADHD.38
Linkage analysis
So far, no linkage studies have been performed looking only at aADHD patients, but two included adults with ADHD and one reported on ADHD symptoms in the population. In the former two, multigenerational pedigrees were investigated. One of them investigated 16 pedigrees from a Colombian Paisa genetic isolate, including a total of 375 individuals (126 cases).94 Fine mapping of regions of suggestive linkage allowed identification of several regions with significant evidence of linkage and a family-specific significant logarithm of odds score on chromosome 8q11.23. Although most of the findings indicated novel ADHD susceptibility loci, regions on 8q11 and 17p11 overlapped with suggestive linkage findings on cADHD.95, 96, 97 In a second study of eight pedigrees (154 family members, 95 cases), several significant linkage regions were found in a combined analysis of all families.98 In addition, family-specific significant findings were also present, some of which (on chromosomes 1, 7, 9, 12, 14 and 16) overlapped with regions earlier implicated in cADHD.96, 97, 99, 100, 101, 102, 103, 104, 105 The study on adult ADHD symptoms in the population investigated sibling pairs (approximately 750) and their family members. Linkage was observed on chromosomes 18q21 and 2p25, and suggestive evidence for aADHD loci was present on chromosomes 3p24 and 8p23.106 Meta-analysis of linkage results derived from seven of nine independent studies (mostly on cADHD) was performed in 2008.107 This analysis revealed one region of significant linkage, on the distal part of chromosome 16q, which contains the CDH13 gene, found nominally associated by GWAS in both cADHD and aADHD (see below), and nine loci with nominal or suggestive evidence of linkage.
Genome-wide association studies
While linkage analysis is mainly suited for the identification of loci having moderate to large effects,108 GWAS can identify common variants increasing the disease risk with only small effects. So far, only one has been performed in aADHD. Although not providing genome-wide significance findings, this DNA pooling-based 500K SNP scan109 identified several potential risk genes and revealed remarkable overlap with findings from GWAS in substance-use disorders. By comparing GWAS results with results from the previously reported high-resolution linkage scan in extended pedigrees,98 several loci harboring ADHD risk genes could be confirmed, including the 16q23.1–24.3 locus containing CDH13. The findings provide support for a common effect of genes coding for cell adhesion/pathfinding molecules (for example, CDH13), regulators of synaptic plasticity (for example, catenin alpha 2 (CTNNA2)) and ion channels or related proteins (for example, voltage-dependent L-type calcium channel α 1D subunit (CACNA1D) and dipeptidyl-peptidase 6 (DPP6)). These pathways show strong overlap with the findings from GWASs in cADHD (for a review see ref. 38).
New developments and initiatives
It appears that the genetic factors underlying ADHD (as well as other psychiatric disorders) are of even smaller effect size than anticipated,110 or are not well covered by current study designs. This has inspired researchers to improve their study designs and to come up with alternative research approaches. The most relevant of these developments for the study of adult ADHD are reviewed below.
Improvement in study designs
If effect sizes of individual genetic factors are small, increasing sample size in genetic studies should substantially improve power for gene finding. This realization has led to an increase in the collaborations between research groups. For aADHD specifically, the International Multicentre persistent ADHD CollaboraTion (IMpACT) was formed in 2007. Including researchers from Europe, the United States and Brazil, this consortium coordinates genetic material of over 3500 well-characterized aADHD cases and approximately 4000 controls. IMpACT has performed meta-analyses of several known candidate genes for ADHD45, 66, 70, 75 (Table 1), thereby providing evidence for differences in the genetic predisposition to persistent ADHD compared with cADHD.
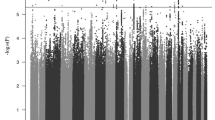
Other examples for collaborative efforts in genetics are the ADHD Molecular Genetics Network,111 a well-established worldwide network of researchers working on ADHD genetics, and the Psychiatric GWAS Consortium (PGC).112 The PGC provides a forum for sharing genome-wide genotyping data and phenotypic information for studies on ADHD (but also on autism spectrum disorder, major depression, bipolar disorder and schizophrenia, see below) (http://www.pgc.unc.edu/index.php).113 Recently, a meta-analysis of ADHD GWASs in the PGC database was performed, including 2064 child–parent trios, 896 cases and 2455 controls.110 This did not yield genome-wide significant findings, yet, potentially due to small effect sizes of individual variants, disease heterogeneity and gene–environment interactions (for a more extensive review of potential reasons for the ‘missing heritability’, see ref. 114). The absence of genome-wide significant findings in a meta-analysis of the current size is not unexpected: a comparison with data on the other disorders within PGC shows a strong correlation between minimal P-values and sample size (Figure 1), suggesting that genome-wide significant findings can only be expected at sample sizes of more than 12 000 individuals (cases and controls combined). These data clearly emphasize the need for multisite collaborations between researchers in ADHD genetics.

Plotted is the sample size (cases+controls) analyzed in the first meta-analyses of the Psychiatric Genome-Wide Association Study (GWAS) Consortium on schizophrenia, bipolar disorder, major depressive disorder, autistic spectrum disorders and attention deficit/hyperactivity disorder (ADHD) against the −log of the minimal association P-value observed in the GWAS. The P-value indicating genome-wide significance of findings is indicated. The data show the strong (r=0.91) and significant (P=0.03) correlation between the two parameters. Drawing a line through the points suggests that at least 12 000 samples (cases+controls) will be needed before genome-wide significant findings for ADHD will be observed.
In such multisite studies of ADHD, one problem is the potential disease heterogeneity among sites due to genetic or cultural differences. However, in this regard it is reassuring that the presentation of ADHD and its prevalence is similar across different countries. Meta-analyses and systematic reviews of epidemiological studies show that there are no differences in the prevalence of ADHD between European countries or between Europe and the United States.115, 116 Similarly, as we reviewed elsewhere,6, 29 the heritability of ADHD does not vary with geographic location. Although these similarities in prevalence and heritability between countries do not assure disease homogeneity, they are consistent with the idea of substantial homogeneity between countries. Furthermore, a large body of literature suggests cross-cultural stability of the ADHD phenotype. Cross-cultural diagnostic studies find no cross-cultural differences in prevalence or expression, when methods of diagnosis are systematized across sites.117 Factor analysis studies have shown that the covariation of ADHD symptoms is invariant across many cultures,118, 119, 120, 121 and cross-cultural studies have also shown considerable stability in the psychiatric and neuropsychological correlates of ADHD.117, 122, 123, 124, 125, 126 In addition to these findings, which were based on the binary diagnosis of ADHD, studies that have used quantitative measures of ADHD show cross-cultural stability in both clinical comorbidity and developmental trends.127, 128, 129, 130 These findings are so compelling that a systematic review of ADHD cross-cultural issues concluded that ‘taken together, these findings suggest that ADHD is not a cultural construct’.131 It is still possible that different sites in a multisite study identify clinically different types of ADHD due to differences in ascertainment (for example, from the population versus clinical samples), exclusion criteria (for example, excluding comorbid disorders) or methodology (for example, the use of different structured diagnostic interviews). The best approach to this problem would be to require sites to use similar methods of ascertainment and assessment.
As mentioned above, the PGC not only brings together data sets for disease-specific GWAS meta-analyses, but also stimulates cross-disorder analyses. This is inspired by the high degree of overlap that has been noted in findings from phenotypic dimensional and molecular genetic studies (for example, refs. 38,132,133); especially, autism and bipolar disorder have a high degree of comorbidity with ADHD, which seems to be caused—at least in part—by overlapping genetic factors.134, 135, 136, 137 Findings from disease-specific GWAS also show association across diagnoses, like the findings that the bipolar risk gene diagylglycerol kinase H (DGKH) is also associated with adult ADHD,138 whereas the ADHD risk gene DIRAS family, GTP-binding RAS-like 2 (DIRAS2), vice versa, is also associated with bipolar disorder.139 Such candidate studies exemplify how common variants might influence disorders on the dimensional, syndromic level—for example, emotional dysregulation—while not being associated with a specific disorder per se. Although this seems plausible for common genetic variants, intuitively one would say that rare variants should be more specific for certain diseases. However, previous examples from studies of rare variants in ADHD, namely copy number variants (CNVs), show an enrichment of CNVs at sites linked to autism and schizophrenia140 (IMAGE II Consortium, under review). Classical approaches relying on traditional nosology fall short to explain such data. As the edge of dawn of more biologically orientated diagnostic systems might be near, the large-scale cross-disorder studies might be one step to elucidate functional genetic networks underlying psychiatric dysfunctioning.
Studies on endophenotype and intermediate phenotypes
Intermediate phenotypes are traits that mediate the association between clinical phenotypes and genes.141, 142 Following the recently suggested terminology from Kendler and Neale,143 endophenotypes reflect measures of brain function that are genetically correlated with a clinical disorder or trait (that is, they share genetic risk factors), whereas the term intermediate phenotype should be reserved for measures that mediate the association between genes and clinical phenotypes. To be considered an endophenotype, a trait must meet several requirements, which include heritability, co-segregation with disease in families, association with disease in the population and higher trait scores in unaffected siblings of patients compared with controls, as well as a criterion relating to the measurement having to be highly accurate and reliable.144 The endophenotypes identified according to these criteria are variables that index genetic risk for disorders, such as ADHD, and include mediating pathways (intermediate phenotypes) as well as pleiotropic phenotypes that reflect multiple different effects of genes. To identify an intermediate phenotype among the endophenotypes requires the additional step of demonstrating mediation between genes and disorder, which can only be tested once one or more genetic markers are found that show association to both the clinical disorder and the endophenotype.135, 143 An example relevant to the study of ADHD is the finding that social cognition mediates the association between the COMT gene and antisocial behavior in cADHD, whereas measures of executive function that were also associated with COMT were found to reflect pleiotropic (multiple outcomes of genes) rather than mediating effects.145
Compared with categorical diagnoses such as ADHD, endophenotypes are assumed to be more proximal to genes in biological pathways (whether they represent intermediate or pleiotropic effects) and to be genetically less complex and giving rise to greater effect sizes of genetic variants. This makes endophenotypes better suited for genetic studies than clinical phenotypes.36, 38 Both endophenotypes and intermediate phenotypes may be used to map genes associated with ADHD, but only intermediate phenotypes can be used to identify the processes that are involved directly in the etiology of ADHD.
Several neurocognitive traits may serve as candidate intermediate phenotypes, because the core features of ADHD (inattention and hyperactivity) are conceptually related with cognitive domains such as executive function, attention, arousal, memory and intelligence.146, 147, 148 Most research in this area has focused on children.149 A meta-analysis of 83 studies involving executive functions (EFs) in cADHD consistently identified deficits on group measures of response inhibition, vigilance, working memory and planning, but noted moderate effect sizes and lack of universality.150 Indeed, (c)ADHD shows considerable heterogeneity with regard to any single cognitive deficit.147 For example, nearly 80% of children with ADHD have a deficit on at least one measure of executive function, but this can also be said of around half of control subjects.151 Temporal processing (response variability),152, 153 visuospatial and verbal working memory,152, 154 response inhibition as measured by the stop-signal reaction time task and interference control149, 154, 155, 156, 157, 158 seem to fulfill the basic criteria for endophenotypes of ADHD and may represent mediating processes. Recently, multivariate analysis of a large cADHD proband, sibling and control sample identified two main familial cognitive factors. The larger factor, which reflected 85% of the familial variance of ADHD, captured all familial influences on response times and response time variability, whereas a second smaller factor reflecting 12.5% of the familial effects on ADHD captured influences on omission errors and commission errors on a go/no-go task.159 These findings may be particularly relevant to aADHD because they reflect two separate developmental processes indexing arousal and attention processes that are hypothesized to underlie persistence and remission of ADHD during the transition into adulthood.160, 161
In aADHD, a range of neurocognitive deficits has also been reported, including problems in sustained attention,162, 163, 164, 165 verbal fluency,166 set shifting,162, 165, 166 word reading,167 color naming,167 verbal and visual working memory,167, 168 interference control165, 169 and response inhibition.163, 166, 170, 171, 172, 173 A meta-analysis of 33 studies concluded that neurocognitive deficits in adults with ADHD are found across a range of domains, in particular involving attention, behavioral inhibition and working memory, with normal performance for simple reaction times.174 Which of these measures satisfy the formal criteria for endophenotypes and intermediate phenotypes of aADHD has yet to be fully investigated.
Despite these promising findings, few neuropsychological phenotypes have yet been used in molecular genetic studies of ADHD, let alone aADHD. There is currently no robust evidence for association between candidate intermediate phenotypes and ADHD candidate genes.175 For aADHD, only two neuropsychological endophenotype studies have been published. Barkley and co-workers41 found association between the 3′-UTR VNTR of the SLC6A3/DAT1 and making more omission errors on a continuous performance test, and the DBH TaqI A2 allele-homozygous participants took more risks in a card playing game. The DRD4 exon 3 VNTR did not have any effects (Table 1). A pilot study in 45 adults with ADHD compared the performance of carriers and non-carriers of ADHD risk alleles in DRD4 (exon 3 VNTR, 120 bp promoter insertion/deletion), SLC6A3/DAT1 (3′-UTR VNTR) and COMT (Val158Met) on a large battery of neurocognitive tests. The study showed COMT to be related to differences in IQ and reaction time, an association of DRD4 with verbal memory skills, and linked SLC6A3/DAT1 to differences in inhibition.176 Two linkage studies reported suggestive loci for traits derived from several neuropsychological tasks.177, 178 With one important prerequisite for endophenotypes suitable for use in genetic studies being measurement errors smaller than those of the related clinical phenotype, single neurocognitive tests may not be the most suitable targets for genetic testing, as they can be prone to several sources of measurement error due to fluctuations in mental state and motivation, stress, fatigue or time of the day.143 A potentially better situation is provided by the use of aggregated measures across neuropsychological tasks in the same way that aggregation of tests is used to estimate IQ. Studies showing the general feasibility of such an approach for gene finding have been performed in children with ADHD (see above; ref. 159).
Structural and functional neuroimaging measures, including both magnetic resonance imaging and cognitive electrophysiology, may be even better suited as endophenotypes, as they generally show strong test–retest reliability in adolescents and adults.179, 180, 181, 182 Two recent meta-analyses suggest that genetic effect sizes at the level of brain activity may be considerable.183, 184 There is ample evidence for dysfunction and subtle structural brain anomalies in ADHD. Most studies have focused on functional aspects of dysfunction reporting deficits in the domains of verbal working memory,185, 186, 187, 188 response inhibition,189, 190 error monitoring191, 192, 193 as well as reward processing and delay aversion.194, 195 Again, only a few studies in aADHD have been published, and there are almost no findings that can be considered replicated (see for a review ref. 196). Studies vary largely by imaging method (functional magnetic resonance imaging or event-related potentials) and paradigm, and almost every research group uses slightly different versions of a given task. In structural imaging, brain volumetry studies in aADHD patients reported reductions of brain volume in the prefrontal cortex196, 197 and anterior cingulate cortex,198 caudate nucleus199, 200 and amygdala,201 as well as a marginal increase of nucleus accumbens volume.196 Only some of these findings have been replicated, to date.200, 202 Interesting recent findings also show structural and functional brain connectivity to be disturbed in ADHD.203, 204
Few studies yet have reported effects of ADHD candidate genes on imaging phenotypes in aADHD. By means of event-related potentials elicited by a go/no-go paradigm and subsequent topographical analysis, it was shown that TPH2 risk alleles previously linked to ADHD205 were associated with reduced no-go anteriorization (suggested to reflect prefrontal brain activity) in aADHD patients as well as healthy controls.206 Likewise, the 9-repeat allele of the SLC6A3/DAT1 3′-UTR VNTR (associated with aADHD45) resulted in a reduction of the no-go anteriorization,207 whereas homozygosity for the 10-repeat allele (which is linked to a higher expression of the transporter in striatum, at least in healthy adults using SPECT208, 209) was associated with hypoactivation in the left dorsal anterior cingulate cortex compared with 9-repeat allele carriership in aADHD patients,210 and a stronger working memory task-related suppression in left medial prefrontal cortex was found in 9-repeat allele carriers compared with 10/10 homozygotes.47 The ADHD risk haplotype of LPHN3 was found associated with no-go anteriorization,211 and was also shown by proton magnetic resonance spectroscopy to decrease the N-acetylaspartate to creatine ratio in the left lateral and medial thalamus and the right striatum, regions altered volumetrically and/or functionally in ADHD.81 Also, the NOS1 exon 1f VNTR showed reduced no-go anteriorization in the controls of a study of impulse-disorder patients (including aADHD patients) homozygous for the short allele of the VNTR, the ADHD risk genotype.212 More recently, an investigation of this variant showed both homozygous short allele aADHD patients and healthy controls to display higher ventral striatal activity during reward anticipation than subjects with the other genotypes.213 A study investigating electroencephalogram measures found an effect of the DRD4 7-repeat allele on the power in the electroencephalogram beta band.214 Furthermore, subjects with this allele were found to have a significantly smaller mean volume in the superior frontal cortex and cerebellum cortex compared with subjects without this allele.215
Based on the above, endophenotypes may be very promising tools for the characterization of biological pathways from gene to disease on the one hand and for gene finding in ADHD on the other. However, as discussed, one should not automatically assume a simple mediational relationship between an endophenotype and a clinical phenotype. Reality may be much more complex. Endophenotypes may be risk indicators of the occurrence or the severity of the clinical phenotype, without exerting a causal influence, genetic influences are expected to be only partially shared between endophenotype and clinical phenotype, and even where mediation is demonstrated, the influences between intermediate phenotype and clinical phenotype could be bi-directional.135, 143 This all complicates the use of endophenotypes in a straightforward way to identify genes for ADHD. Moreover, as has been shown in research of autism, similar genetic variants may influence a very broad range of endophenotypes, suggesting that the effective distance between variations in the sequence or structure of the DNA and resulting brain endophenotypes may be still quite large.216 Nonetheless, using endophenotypes continues to be a powerful way to unravel the genetic architecture of multifactorial disorders such as aADHD, but its effective application may require moving to more comprehensive approaches that include the simultaneous modeling of multiple endophenotypes, innovative statistical methods and the combination of those with bioinformatics.217 Assessing endophenotypes in multisite collaborative studies would additionally require prospective studies using identical measures across sites, or the definition of derived (aggregate) measures that capture the underlying trait optimally while reducing task-specific measurement noise (for example, ref. 159).
New methods for the statistical analysis of genetic association
Hypothesis-driven candidate gene studies have been the focus of many research groups, as, with current sample sizes, they provide superior power. However, instead of investigating single polymorphisms, entire genes or even entire functional networks are currently being investigated. The first examples of these studies have focused on the association of neurotrophic factors,90, 74 the serotonergic system68 and brain laterality-related genes73 with aADHD (see Table 1). Tools like the KEGG Pathway Database (http://www.genome.jp/kegg/pathway.html), Gene Ontology (http://www.geneontology.org) or DAVID (http://www.david.abcc.ncifcrf.gov) are useful for identifying possible candidate systems and selecting the constituent genes. This approach may also be applied to the analysis of data from genome-wide genotyping efforts, by calculating association scores between the disorder and functional groups. An example of a statistical approach for this was recently published for the analysis of IQ in a sample of children with ADHD,218 but similar univariate as well as multivariate approaches have been suggested.219, 220, 221, 222
With the improvement of statistical methods, the investigation of gene-by-environment (G × E) and gene-by-gene (G × G) interactions is becoming more and more feasible. So far, only very few studies have addressed this issue in aADHD (Table1 and above), in largely underpowered studies. In cADHD, more literature is available, but results for individual genes are still conflicting.223, 224, 225
Bioinformatic analyses are becoming more and more important as a tool for the integration of genetic findings. Such analyses can indicate biological processes and pathways enriched in the data from GWASs.226 In ADHD, a study on copy number variants (see below) showed enrichment for genes important for learning, behavior, synaptic transmission and central nervous system development227 using bioinformatics. Another recent study integrated the top-ranked findings of all published GWAS in ADHD and found a strong enrichment of genes related to neurite outgrowth.228
Investigation of rare genetic causes of ADHD and alternative patterns of genetic transmission
Judging from the high prevalence of ADHD in the general population and the strong decline of disease risk from first- to second-degree relatives, a multifactorial polygenic inheritance model has been considered most likely for ADHD.229, 230With the involvement of environmental factors, the disorder seems best described as being of multifactorial origin. The multifactorial polygenic model has motivated the search for common DNA variants, as described above, using candidate gene, genome-wide linkage and GWAS. However, given the limited success of GWAS in ADHD, thus far,110, 231 in conjunction with reports on increased burden of rare copy number variants in, for example, schizophrenia and autism (for example, refs. 232,233), ADHD researchers have begun the search for rare variants that might account for some of ADHD's heritability.
From case reports, we have known for a long time that unique mutations can lead to ADHD. Examples include a translocation involving the solute carrier family 9 member 9 gene, SLC9A9,234 and an inactivating mutation in TPH2,235 both found to co-segregate with ADHD in two different families, but also larger chromosomal abnormalities.236, 237, 238, 239, 240, 241 In addition, several syndromes caused by rare genetic mutations (including the 22q11 deletion syndrome and Klinefelter syndrome) are known to show increased incidence of ADHD(-like phenotypes),242, 243 although adult forms of ADHD are often not part of the clinical assessment of these patients.
Most of the earlier studies have not systematically investigated the entire genome for rare, deleterious mutations, nor did they indicate whether such mutations also cause ADHD in adults. A first systematic analysis of microdeletions and duplications (CNVs), including adults with ADHD, has been published recently.244 This study revealed de novo as well as inherited CNVs associated with ADHD. A particularly interesting finding from this study includes an extended pedigree with multiple cases of ADHD and obesity, in which a duplication of the gene encoding neuropeptide Y (NPY) was observed. From this, in conjunction with a number of studies systematically investigating CNVs in data from GWASs of cADHD (published,227, 245, 246 or currently under review), it becomes clear that some ADHD cases—rather than being caused by multiple common variants—may be caused by rare genetic variants with relatively large effect sizes. What fraction of ADHD cases can be explained by such oligogenic (or perhaps even monogenic) causes, however, will have to await studies involving genome-wide sequencing,247, 248 as microdeletions and duplications are likely to be not the only type of genetic variant involved. The study of extended pedigrees with multiple affected members might provide a shortcut to finding some of the altered genes. Intriguingly, a recent publication also suggests that some associations found in GWAS studies—seemingly caused by common variants—might actually be based on synthetic association with rare variants in partial linkage disequilibrium with the common variants.249
In addition to considering rare versus common genetic variants, alternative patterns of genetic transmission should be considered. Based on studies of rare coding variants affecting the function of TPH1, a strong maternal transmission of the risk allele was suggested.250 Parent of origin effects have also been suggested for common variants in cADHD.251, 252 Similar specific patterns of genetic transmission could occur for many candidate genes, but the effects would easily be obscured in case–control studies.
Clinical impact of understanding the genetics of adult ADHD
Compared with other clinical neurosciences, little progress has been made in the application of molecular diagnostics to the common psychiatric disorders. Notably, genetic tests are now commonly being used in the diagnosis of early-onset neurodegenerative disorders. Although Parkinson's disease has traditionally been considered a non-genetic disease, during the past 15 years many rare Mendelian and common low-risk loci for this disease have been successfully identified. Taken together, these loci account for about half of the accumulated risk of developing early-onset Parkinson's disease and genetic testing of these markers has rapidly become useful for diagnosis and for defining new therapeutic strategies.253 In analogy with such examples, it may be possible to identify susceptibility genes in subgroups of patients with monogenic or oligogenic forms of aADHD by sequencing and genotyping pedigrees with a high load of this disorder. Genotyping of such rare, highly penetrant genetic variants may have clinical utility where aADHD needs to be differentiated from progressive neurological conditions or other somatic or psychiatric disorders. Although our current understanding of the genetic models of transmission and the variants involved is still limited, with increasing knowledge of these variants, and in the hands of experts in psychiatric genetics, this might become feasible in the future. However, as the susceptibility genes that have been robustly identified in GWASs of psychiatric disorders so far seem to confer vulnerability across a range of psychiatric phenotypes and the genetic markers have very low predictive value,38, 133, 254, 255 it is expected that such a clinical application of aADHD genetics will only appear gradually.
Another way of incorporating the results of genetic research into clinical practice is pharmacogenetics, the individualization of treatment strategies based on the association of DNA variants with drug efficacy or adverse events. Pharmacogenetic testing may be able to help clinicians in individualizing the treatment option for any ADHD patient, in terms of efficacy and tolerability.256, 257 In all, 30% of aADHD patients do not respond favorably to stimulant treatment (methylphenidate or amphetamines) and 40% exhibit non-response to atomoxetine. In addition, many patients present side effects with these drugs, like an increase in arterial tension or insomnia, that can cause them to drop out of treatment.258 However, efforts at understanding the putative role of candidate genes in the response to pharmacotherapy for ADHD have been inconclusive,259 a pharmacogenetic GWAS found no genome-wide significant associations,260 and—with the exception of a few studies261, 262, 263—the pertinent literature is exclusively focused on pediatric samples and on a few genes.
Prediction of outcome and prevention of persistence through intervention is a particularly relevant clinical issue. Knowing that ADHD remits in a percentage of cases,2 and that both genetic and environmental factors are involved in its etiology provides a basis for hypothesizing that ADHD persistence into adulthood might be preventable in some patients by intervention early in childhood. Indeed, the finding from longitudinal twin studies of ADHD throughout child and adolescent development suggest a role for newly developing genetic influences at different developmental states,9, 264 but further twin studies are needed that span from adolescence through into adulthood. The literature on the prognostic value of individual genetic factors is still contradictory. In this regard, ADHD children carrying the DRD4 7-repeat allele show normalization of the cortical thinning in the right parietal cortical region, a pattern that linked with better clinical outcome.86 In contrast, others showed that in ADHD patients reassessed after 5 years, carriers of the DRD4 7-repeat allele showed less decline in severity than those without the risk allele.265 Other findings indicate that DRD4 7-repeat allele carriers are more persistently affected than those not carrying this risk allele,48 and no effect of DRD4 was observed in another study.41 A meta-analysis of SLC6A3/DAT1 by IMpACT suggests that a different haplotype from that reported associated with cADHD is associated with aADHD,44, 45 see above. In line with this, carriers of the 9/10 genotype of the 3′-UTR VNTR were earlier shown to have a worse prognosis than those with the 10/10 genotype.41 Additional genetic analyses in large longitudinal studies will be needed to investigate (patterns of) genetic variants of potential value.
Looking forward
In this paper, we critically reviewed current literature on the genetics of aADHD, the most severe form of the disorder. So far, this is still limited, as most work has been concentrated on the disorder in children.
The extent of heritability of ADHD in adults has not been firmly established, and stringently characterized samples should be used to provide more exact estimates.
Adult ADHD etiology is likely to involve both common and rare genetic variants. Although the search for common DNA variants predisposing for ADHD has not yet successfully achieved the level of genome-wide significance, recently reported genome-wide significant effects for other psychiatric disorders (for example, ref. 266) suggest that similar findings for ADHD will be forthcoming. Given that the effects of common genetic variants are expected to be very small, their relevance to ADHD cannot be ruled out in currently available samples. Therefore, more genome-wide studies of common as well as rare variants are absolutely necessary. In addition, we should strive for improvements in the statistical tools used to perform such studies as well as those enabling integration of the findings.
Circumventing clinical heterogeneity, endophenotypes based on neuroimaging and multivariate measures of neuropsychology can help to identify new genes for aADHD. In addition, such brain phenotypes can provide more insight into the mechanisms underlying disease etiology by enabling the mapping of biological pathways from gene to disease.
First indications suggest that the genetic component of aADHD is partly different from the one observed in children, which may leave room for differentiating persisters from desisters in the future. Given this prospect, in conjunction with the prospects of using genetics in the clinic to improve treatment for ADHD in adults, halt the progression of the disorder and/or improve coping when the disorder does persist, large-scale studies of aADHD (genetics), especially those with longitudinal designs, seem warranted.
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th edn, revised. American Psychiatric Press: Washington, DC 2000.
Faraone SV, Biederman J, Mick E . The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med 2006; 36: 159–165.
Simon V, Czobor P, Balint S, Meszaros A, Bitter I . Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry 2009; 194: 204–211.
Barkley RA, Murphy KR, Fischer M . ADHD in Adults: What the Science Says. Guilford Press: New York, NY, 2008.
Haavik J, Halmoy A, Lundervold AJ, Fasmer OB . Clinical assessment and diagnosis of adults with attention-deficit/hyperactivity disorder. Expert Rev Neurother 2010; 10: 1569–1580.
Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 2005; 57: 1313–1323.
Burt SA . Rethinking environmental contributions to child and adolescent psychopathology: a meta-analysis of shared environmental influences. Psychol Bull 2009; 135: 608–637.
van den Berg SM, Willemsen G, de Geus EJ, Boomsma DI . Genetic etiology of stability of attention problems in young adulthood. Am J Med Genet B 2006; 141B: 55–60.
Larsson JO, Larsson H, Lichtenstein P . Genetic and environmental contributions to stability and change of ADHD symptoms between 8 and 13 years of age: a longitudinal twin study. J Am Acad Child Adolesc Psychiatry 2004; 43: 1267–1275.
Kuntsi J, Rijsdijk F, Ronald A, Asherson P, Plomin R . Genetic influences on the stability of attention-deficit/hyperactivity disorder symptoms from early to middle childhood. Biol Psychiatry 2005; 57: 647–654.
Morrison JR, Stewart MA . A family study of the hyperactive child syndrome. Biol Psychiatry 1971; 3: 189–195.
Cantwell DP . Psychiatric illness in the families of hyperactive children. Arch Gen Psychiatry 1972; 27: 414–417.
Biederman J, Faraone SV, Keenan K, Knee D, Tsuang MT . Family-genetic and psychosocial risk factors in DSM-III attention deficit disorder. J Am Acad Child Adolesc Psychiatry 1990; 29: 526–533.
Faraone SV, Biederman J, Keenan K, Tsuang MT . Separation of DSM-III attention deficit disorder and conduct disorder: evidence from a family-genetic study of American child psychiatric patients. Psychol Med 1991; 21: 109–121.
Biederman J, Faraone SV, Mick E, Spencer T, Wilens T, Kiely K et al. High risk for attention deficit hyperactivity disorder among children of parents with childhood onset of the disorder: a pilot study. Am J Psychiatry 1995; 152: 431–435.
Manshadi M, Lippmann S, O’Daniel RG, Blackman A . Alcohol abuse and attention deficit disorder. J Clin Psychiatry 1983; 44: 379–380.
Biederman J, Faraone S, Milberger S, Curtis S, Chen L, Marrs A et al. Predictors of persistence and remission of ADHD into adolescence: results from a four-year prospective follow-up study. J Am Acad Child Adolesc Psychiatry 1996; 35: 343–351.
Faraone SV, Biederman J, Monuteaux MC . Toward guidelines for pedigree selection in genetic studies of attention deficit hyperactivity disorder. Genet Epidemiol 2000; 18: 1–16.
Alberts-Corush J, Firestone P, Goodman JT . Attention and impulsivity characteristics of the biological and adoptive parents of hyperactive and normal control children. Am J Orthopsychiatry 1986; 56: 413–423.
Sprich S, Biederman J, Crawford MH, Mundy E, Faraone SV . Adoptive and biological families of children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry 2000; 39: 1432–1437.
Epstein JN, Conners CK, Erhardt D, Arnold LE, Hechtman L, Hinshaw SP et al. Familial aggregation of ADHD characteristics. J Abnorm Child Psychol 2000; 28: 585–594.
Epstein J, Johnson DE, Conners CK (ed). Conners’ Adult ADHD Diagnostic Interview for DSM-IV (CAADID). Multi-Health Systems: North Tonawanda, NY, 2001.
Curko Kera EA, Marks DJ, Berwid OG, Santra A, Halperin JM . Self-report and objective measures of ADHD-related behaviors in parents of preschool children at risk for ADHD. CNS Spectr 2004; 9: 639–647.
Schultz MR, Rabi K, Faraone SV, Kremen W, Lyons MJ . Efficacy of retrospective recall of attention-deficit hyperactivity disorder symptoms: a twin study. Twin Res Hum Genet 2006; 9: 220–232.
Boomsma DI, Saviouk V, Hottenga JJ, Distel MA, de Moor MH, Vink JM . et al. Genetic epidemiology of attention deficit hyperactivity disorder (ADHD index) in adults. PLoS One 2010; 5: e10621.
Martin N, Scourfield J, McGuffin P . Observer effects and heritability of childhood attention-deficit hyperactivity disorder symptoms. Br J Psychiatry 2002; 180: 260–265.
Ehringer MA, Rhee SH, Young S, Corley R, Hewitt JK . Genetic and environmental contributions to common psychopathologies of childhood and adolescence: a study of twins and their siblings. J Abnorm Child Psychol 2006; 34: 1–17.
McLoughlin G, Ronald A, Kuntsi J, Asherson P, Plomin R . Genetic support for the dual nature of attention deficit hyperactivity disorder: substantial genetic overlap between the inattentive and hyperactive-impulsive components. J Abnorm Child Psychol 2007; 35: 999–1008.
Faraone SV, Mick E . Molecular genetics of attention deficit hyperactivity disorder. Psychiatr Clin N Am 2010; 33: 159–180.
Kessler RC, Adler L, Ames M, Demler O, Faraone S, Hiripi E et al. The World Health Organization Adult ADHD Self-Report Scale (ASRS): a short screening scale for use in the general population. Psychol Med 2005; 35: 245–256.
Daigre BC, Ramos-Quiroga JA, Valero S, Bosch R, Roncero C, Gonzalvo B et al. Adult ADHD Self-Report Scale (ASRS-v1.1) symptom checklist in patients with substance use disorders. Actas Esp Psiquiatr 2009; 37: 299–305.
Biederman J, Mick E, Faraone SV . Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am J Psychiatry 2000; 157: 816–818.
Faraone SV . Attention deficit hyperactivity disorder in adults: implications for theories of diagnosis. Curr Direct Psychol Sci 2000; 9: 33–36.
Schultz MR, Rabi K, Faraone SV, Kremen W, Lyons MJ . Efficacy of retrospective recall of attention-deficit hyperactivity disorder symptoms: A twin study. Twin Res Hum Genet 2006; 9: 220–232.
Kendler KS, Myers J, Torgersen S, Neale MC, Reichborn-Kjennerud T . The heritability of cluster A personality disorders assessed by both personal interview and questionnaire. Psychol Med 2007; 37: 655–665.
Flint J, Munafo MR . The endophenotype concept in psychiatric genetics. Psychol Med 2007; 37: 163–180.
Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet 2010; 42: 570–575.
Franke B, Neale BM, Faraone SV . Genome-wide association studies in ADHD. Hum Genet 2009; 126: 13–50.
Gizer IR, Ficks C, Waldman ID . Candidate gene studies of ADHD: a meta-analytic review. Hum Genet 2009; 126: 51–90.
Muglia P, Jain U, Inkster B, Kennedy JL . A quantitative trait locus analysis of the dopamine transporter gene in adults with ADHD. Neuropsychopharmacology 2002; 27: 655–662.
Barkley RA, Smith KM, Fischer M, Navia B . An examination of the behavioral and neuropsychological correlates of three ADHD candidate gene polymorphisms (DRD4 7+, DBH TaqI A2, and DAT1 40 bp VNTR) in hyperactive and normal children followed to adulthood. Am J Med Genet B 2006; 141: 487–498.
Bruggemann D, Sobanski E, Alm B, Schubert T, Schmalzried H, Philipsen A et al. No association between a common haplotype of the 6 and 10-repeat alleles in intron 8 and the 3′UTR of the DAT1 gene and adult attention deficit hyperactivity disorder. Psychiatr Genet 2007; 17: 121.
Johansson S, Halleland H, Halmoy A, Jacobsen KK, Landaas ET, Dramsdahl M et al. Genetic analyses of dopamine related genes in adult ADHD patients suggest an association with the DRD5-microsatellite repeat, but not with DRD4 or SLC6A3 VNTRs. Am J Med Genet B 2008; 147B: 1470–1475.
Franke B, Hoogman M, Arias VA, Heister JG, Savelkoul PJ, Naber M et al. Association of the dopamine transporter (SLC6A3/DAT1) gene 9-6 haplotype with adult ADHD. Am J Med Genet B 2008; 147B: 1576–1579.
Franke B, Vasquez AA, Johansson S, Hoogman M, Romanos J, Boreatti-Hummer A et al. Multicenter analysis of the SLC6A3/DAT1 VNTR haplotype in persistent ADHD suggests differential involvement of the gene in childhood and persistent ADHD. Neuropsychopharmacology 2010; 35: 656–664.
da Silva MA, Cordeiro Q, Louza M, Vallada H . Lack of association between a 3′UTR VNTR polymorphism of dopamine transporter gene (SLC6A3) and ADHD in a Brazilian sample of adult patients. J Atten Disord 2011; 15: 305–309.
Brown AB, Biederman J, Valera E, Makris N, Doyle A, Whitfield-Gabrieli S et al. Relationship of DAT1 and adult ADHD to task-positive and task-negative working memory networks. Psychiatry Res 2011; 193: 7–16.
Biederman J, Petty CR, Ten Haagen KS, Small J, Doyle AE, Spencer T et al. Effect of candidate gene polymorphisms on the course of attention deficit hyperactivity disorder. Psychiatry Res 2009; 170: 199–203.
Muller DJ, Chiesa A, Mandelli L, De LV, De RD, Jain U et al. Correlation of a set of gene variants, life events and personality features on adult ADHD severity. J Psychiatr Res 2010; 44: 598–604.
Sanchez-Mora C, Ribases M, Casas M, Bayes M, Bosch R, Fernandez-Castillo N et al. Exploring DRD4 and its interaction with SLC6A3 as possible risk factors for adult ADHD: a meta-analysis in four European populations. Am J Med Genet B 2011; 156: 600–612.
Muglia P, Jain U, Macciardi F, Kennedy JL . Adult attention deficit hyperactivity disorder and the dopamine D4 receptor gene. Am J Med Genet 2000; 96: 273–277.
Arcos-Burgos M, Castellanos FX, Konecki D, Lopera F, Pineda D, Palacio JD et al. Pedigree disequilibrium test (PDT) replicates association and linkage between DRD4 and ADHD in multigenerational and extended pedigrees from a genetic isolate. Mol Psychiatry 2004; 9: 252–259.
Lynn DE, Lubke G, Yang M, McCracken JT, McGough JJ, Ishii J et al. Temperament and character profiles and the dopamine D4 receptor gene in ADHD. Am J Psychiatry 2005; 162: 906–913.
Muglia P, Jain U, Kennedy JL . A transmission disequilibrium test of the Ser9/Gly dopamine D3 receptor gene polymorphism in adult attention-deficit hyperactivity disorder. Behav Brain Res 2002; 130: 91–95.
Davis C, Patte K, Levitan RD, Carter J, Kaplan AS, Zai C et al. A psycho-genetic study of associations between the symptoms of binge eating disorder and those of attention deficit (hyperactivity) disorder. J Psychiatr Res 2009; 43: 687–696.
Kim JW, Park CS, Hwang JW, Shin MS, Hong KE, Cho SC et al. Clinical and genetic characteristics of Korean male alcoholics with and without attention deficit hyperactivity disorder. Alcohol Alcohol 2006; 41: 407–411.
Sizoo B, van den BW, Franke B, Vasquez AA, van Wijngaarden-Cremers P, van der Gaag RJ . Do candidate genes discriminate patients with an autism spectrum disorder from those with attention deficit/hyperactivity disorder and is there an effect of lifetime substance use disorders? World J Biol Psychiatry 2010; 11: 699–708.
Squassina A, Lanktree M, De LV, Jain U, Krinsky M, Kennedy JL et al. Investigation of the dopamine D5 receptor gene (DRD5) in adult attention deficit hyperactivity disorder. Neurosci Lett 2008; 432: 50–53.
Inkster B, Muglia P, Jain U, Kennedy JL . Linkage disequilibrium analysis of the dopamine beta-hydroxylase gene in persistent attention deficit hyperactivity disorder. Psychiatr Genet 2004; 14: 117–120.
Hess C, Reif A, Strobel A, Boreatti-Hummer A, Heine M, Lesch KP et al. A functional dopamine-beta-hydroxylase gene promoter polymorphism is associated with impulsive personality styles, but not with affective disorders. J Neural Transm 2009; 116: 121–130.
Muller DJ, Mandelli L, Serretti A, DeYoung CG, De LV, Sicard T et al. Serotonin transporter gene and adverse life events in adult ADHD. Am J Med Genet B 2008; 147B: 1461–1469.
Retz W, Rosler M, Kissling C, Wiemann S, Hunnerkopf R, Coogan A et al. Norepinephrine transporter and catecholamine-O-methyltransferase gene variants and attention-deficit/hyperactivity disorder symptoms in adults. J Neural Transm 2008; 115: 323–329.
Halleland H, Lundervold AJ, Halmoy A, Haavik J, Johansson S . Association between catechol O-methyltransferase (COMT) haplotypes and severity of hyperactivity symptoms in adults. Am J Med Genet B 2009; 150B: 403–410.
Johann M, Bobbe G, Putzhammer A, Wodarz N . Comorbidity of alcohol dependence with attention-deficit hyperactivity disorder: differences in phenotype with increased severity of the substance disorder, but not in genotype (serotonin transporter and 5-hydroxytryptamine-2c receptor). Alcohol Clin Exp Res 2003; 27: 1527–1534.
Grevet EH, Marques FZ, Salgado CA, Fischer AG, Kalil KL, Victor MM et al. Serotonin transporter gene polymorphism and the phenotypic heterogeneity of adult ADHD. J Neural Transm 2007; 114: 1631–1636.
Landaas ET, Johansson S, Jacobsen KK, Ribases M, Bosch R, Sanchez-Mora C et al. An international multicenter association study of the serotonin transporter gene in persistent ADHD. Genes Brain Behav 2010; 9: 449–458.
Jacob CP, Nguyen TT, Dempfle A, Heine M, Windemuth-Kieselbach C, Baumann K et al. A gene–environment investigation on personality traits in two independent clinical sets of adult patients with personality disorder and attention deficit/hyperactive disorder. Eur Arch Psychiatry Clin Neurosci 2010; 260: 317–326.
Ribases M, Ramos-Quiroga JA, Hervas A, Bosch R, Bielsa A, Gastaminza X et al. Exploration of 19 serotoninergic candidate genes in adults and children with attention-deficit/hyperactivity disorder identifies association for 5HT2A, DDC and MAOB. Mol Psychiatry 2009; 14: 71–85.
Reuter M, Kirsch P, Hennig J . Inferring candidate genes for attention deficit hyperactivity disorder (ADHD) assessed by the World Health Organization Adult ADHD Self-Report Scale (ASRS). J Neural Transm 2006; 113: 929–938.
Johansson S, Halmoy A, Mavroconstanti T, Jacobsen KK, Landaas ET, Reif A et al. Common variants in the TPH1 and TPH2 regions are not associated with persistent ADHD in a combined sample of 1,636 adult cases and 1,923 controls from four European populations. Am J Med Genet B 2010; 153B: 1008–1015.
De Luca V, Muglia P, Vincent JB, Lanktree M, Jain U, Kennedy JL . Adrenergic alpha 2C receptor genomic organization: association study in adult ADHD. Am J Med Genet B 2004; 127B: 65–67.
de Cerqueira CC, Polina ER, Contini V, Marques FZ, Grevet EH, Salgado CA et al. ADRA2A polymorphisms and ADHD in adults: possible mediating effect of personality. Psychiatry Res 2011; 186: 345–350.
Ribases M, Bosch R, Hervas A, Ramos-Quiroga JA, Sanchez-Mora C, Bielsa A et al. Case–control study of six genes asymmetrically expressed in the two cerebral hemispheres: association of BAIAP2 with attention-deficit/hyperactivity disorder. Biol Psychiatry 2009; 66: 926–934.
Conner AC, Kissling C, Hodges E, Hunnerkopf R, Clement RM, Dudley E et al. Neurotrophic factor-related gene polymorphisms and adult attention deficit hyperactivity disorder (ADHD) score in a high-risk male population. Am J Med Genet B 2008; 147B: 1476–1480.
Sanchez-Mora C, Ribases M, Ramos-Quiroga JA, Casas M, Bosch R, Boreatti-Hummer A et al. Meta-analysis of brain-derived neurotrophic factor p.Val66Met in adult ADHD in four European populations. Am J Med Genet B 2010; 153B: 512–523.
Lanktree M, Squassina A, Krinsky M, Strauss J, Jain U, Macciardi F et al. Association study of brain-derived neurotrophic factor (BDNF) and LIN-7 homolog (LIN-7) genes with adult attention-deficit/hyperactivity disorder. Am J Med Genet B 2008; 147B: 945–951.
De Luca V, Muglia P, Jain U, Basile VS, Sokolowski MB, Kennedy JL . A Drosophila model for attention deficit hyperactivity disorder (ADHD): No evidence of association with PRKG1 gene. Neuromolecular Med 2002; 2: 281–287.
Kissling C, Retz W, Wiemann S, Coogan AN, Clement RM, Hunnerkopf R et al. A polymorphism at the 3′-untranslated region of the CLOCK gene is associated with adult attention-deficit hyperactivity disorder. Am J Med Genet B 2008; 147: 333–338.
Lu AT, Ogdie MN, Jarvelin MR, Moilanen IK, Loo SK, McCracken JT et al. Association of the cannabinoid receptor gene (CNR1) with ADHD and post-traumatic stress disorder. Am J Med Genet B 2008; 147B: 1488–1494.
Reif A, Jacob CP, Rujescu D, Herterich S, Lang S, Gutknecht L et al. Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch Gen Psychiatry 2009; 66: 41–50.
Arcos-Burgos M, Jain M, Acosta MT, Shively S, Stanescu H, Wallis D et al. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol Psychiatry 2010; 15: 1053–1066.
Ribases M, Ramos-Quiroga JA, Sanchez-Mora C, Bosch R, Richarte V, Alvarez I et al. Contribution of Latrophilin 3 (LPHN3) to the genetic susceptibility to ADHD in adulthood: a replication study. Genes Brain Behav 2011; 10: 149–157.
Landaas ET, Johansson S, Halmoy A, Oedegaard KJ, Fasmer OB, Haavik J . Bipolar disorder risk alleles in adult ADHD patients. Genes Brain Behav 2011; 10: 418–423.
Asherson P, Brookes K, Franke B, Chen W, Gill M, Ebstein RP et al. Confirmation that a specific haplotype of the dopamine transporter gene is associated with combined-type ADHD. Am J Psychiatry 2007; 164: 674–677.
Brookes KJ, Mill J, Guindalini C, Curran S, Xu X, Knight J et al. A common haplotype of the dopamine transporter gene associated with attention-deficit/hyperactivity disorder and interacting with maternal use of alcohol during pregnancy. Arch Gen Psychiatry 2006; 63: 74–81.
Shaw P, Gornick M, Lerch J, Addington A, Seal J, Greenstein D et al. Polymorphisms of the dopamine D4 receptor, clinical outcome, and cortical structure in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 2007; 64: 921–931.
Retz W, Freitag CM, Retz-Junginger P, Wenzler D, Schneider M, Kissling C et al. A functional serotonin transporter promoter gene polymorphism increases ADHD symptoms in delinquents: interaction with adverse childhood environment. Psychiatry Res 2008; 158: 123–131.
De Luca V, Muglia P, Jain U, Kennedy JL . No evidence of linkage or association between the norepinephrine transporter (NET) gene MnlI polymorphism and adult ADHD. Am J Med Genet B 2004; 124B: 38–40.
Ribases M, Hervas A, Ramos-Quiroga JA, Bosch R, Bielsa A, Gastaminza X et al. Association study of 10 genes encoding neurotrophic factors and their receptors in adult and child attention-deficit/hyperactivity disorder. Biol Psychiatry 2008; 63: 935–945.
Ribases M, Hervas A, Ramos-Quiroga JA, Bosch R, Bielsa A, Gastaminza X et al. Association study of 10 genes encoding neurotrophic factors and their receptors in adult and child attention-deficit/hyperactivity disorder. Biol Psychiatry 2008; 63: 935–945.
Martinez AF, Muenke M, Arcos-Burgos M . From the black widow spider to human behavior: Latrophilins, a relatively unknown class of G protein-coupled receptors, are implicated in psychiatric disorders. Am J Med Genet B 2011; 156B: 1–10.
Hale TS, Loo SK, Zaidel E, Hanada G, Macion J, Smalley SL . Rethinking a right hemisphere deficit in ADHD. J Atten Disord 2009; 13: 3–17.
Van Veen MM, Kooij JJ, Boonstra AM, Gordijn MC, Van Someren EJ . Delayed circadian rhythm in adults with attention-deficit/hyperactivity disorder and chronic sleep-onset insomnia. Biol Psychiatry 2010; 67: 1091–1096.
Arcos-Burgos M, Castellanos FX, Pineda D, Lopera F, Palacio JD, Palacio LG et al. Attention-deficit/hyperactivity disorder in a population isolate: linkage to loci at 4q13.2, 5q33.3, 11q22, and 17p11. Am J Hum Genet 2004; 75: 998–1014.
Faraone SV, Doyle AE, Lasky-Su J, Sklar PB, D’Angelo E, Gonzalez-Heydrich J et al. Linkage analysis of attention deficit hyperactivity disorder. Am J Med Genet B 2008; 147B: 1387–1391.
Ogdie MN, Fisher SE, Yang M, Ishii J, Francks C, Loo SK et al. Attention deficit hyperactivity disorder: fine mapping supports linkage to 5p13, 6q12, 16p13, and 17p11. Am J Hum Genet 2004; 75: 661–668.
Ogdie MN, Macphie IL, Minassian SL, Yang M, Fisher SE, Francks C et al. A genomewide scan for attention-deficit/hyperactivity disorder in an extended sample: suggestive linkage on 17p11. Am J Hum Genet 2003; 72: 1268–1279.
Romanos M, Freitag C, Jacob C, Craig DW, Dempfle A, Nguyen TT et al. Genome-wide linkage analysis of ADHD using high-density SNP arrays: novel loci at 5q13.1 and 14q12. Mol Psychiatry 2008; 13: 522–530.
Fisher SE, Francks C, McCracken JT, McGough JJ, Marlow AJ, Macphie IL et al. A genomewide scan for loci involved in attention-deficit/hyperactivity disorder. Am J Hum Genet 2002; 70: 1183–1196.
Ogdie MN, Bakker SC, Fisher SE, Francks C, Yang MH, Cantor RM et al. Pooled genome-wide linkage data on 424 ADHD ASPs suggests genetic heterogeneity and a common risk locus at 5p13. Mol Psychiatry 2006; 11: 5–8.
Bakker SC, van der Meulen EM, Buitelaar JK, Sandkuijl LA, Pauls DL, Monsuur AJ et al. A whole-genome scan in 164 Dutch sib pairs with attention-deficit/hyperactivity disorder: suggestive evidence for linkage on chromosomes 7p and 15q. Am J Hum Genet 2003; 72: 1251–1260.
Hebebrand J, Dempfle A, Saar K, Thiele H, Herpertz-Dahlmann B, Linder M et al. A genome-wide scan for attention-deficit/hyperactivity disorder in 155 German sib-pairs. Mol Psychiatry 2006; 11: 196–205.
Asherson P, Zhou K, Anney RJ, Franke B, Buitelaar J, Ebstein R et al. A high-density SNP linkage scan with 142 combined subtype ADHD sib pairs identifies linkage regions on chromosomes 9 and 16. Mol Psychiatry 2008; 13: 514–521.
Zhou K, Asherson P, Sham P, Franke B, Anney RJ, Buitelaar J et al. Linkage to chromosome 1p36 for attention-deficit/hyperactivity disorder traits in school and home settings. Biol Psychiatry 2008; 64: 571–576.
Vegt R, Bertoli-Avella AM, Tulen JH, de GB, Verkerk AJ, Vervoort J et al. Genome-wide linkage analysis in a Dutch multigenerational family with attention deficit hyperactivity disorder. Eur J Hum Genet 2010; 18: 206–211.
Saviouk V, Hottenga JJ, Slagboom EP, Distel MA, de Geus EJ, Willemsen G et al. ADHD in Dutch adults: heritability and linkage study. Am J Med Genet B 2011; 156B: 352–362.
Zhou K, Dempfle A, Arcos-Burgos M, Bakker SC, Banaschewski T, Biederman J et al. Meta-analysis of genome-wide linkage scans of attention deficit hyperactivity disorder. Am J Med Genet B 2008; 147B: 1392–1398.
Risch N, Merikangas K . The future of genetic studies of complex human diseases. Science 1996; 273: 1516–1517.
Lesch KP, Timmesfeld N, Renner TJ, Halperin R, Roser C, Nguyen TT et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm 2008; 115: 1573–1585.
Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 884–897.
Faraone SV . Report from the 4th international meeting of the attention deficit hyperactivity disorder molecular genetics network. Am J Med Genet B 2003; 121: 55–59.
Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, Kelsoe J et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009; 166: 540–556.
Psychiatric GWAS Consortium Steering Committee. A framework for interpreting genome-wide association studies of psychiatric disorders. Mol Psychiatry 2009; 14: 10–17.
Maher B . Personal genomes: the case of the missing heritability. Nature 2008; 456: 18–21.
Faraone SV, Sergeant J, Gillberg C, Biederman J . The worldwide prevalence of ADHD: is it an American condition? World Psychiatry 2003; 2: 104–113.
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA . The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007; 164: 942–948.
Prendergast M, Taylor E, Rapoport JL, Bartko J, Donnelly M, Zametkin A et al. The diagnosis of childhood hyperactivity. A US–U.K. cross-national study of DSM-III and ICD-9. J Child Psychol Psychiatry 1988; 29: 289–300.
Bauermeister JJ, Alegria M, Bird HR, Rubio-Stipec M, Canino G . Are attentional-hyperactivity deficits unidimensional or multidimensional syndromes? Empirical findings from a community survey. J Am Acad Child Adolesc Psychiatry 1992; 31: 423–431.
Beiser M, Dion R, Gotowiec A . The structure of attention-deficit and hyperactivity symptoms among native and non-native elementary school children. J Abnorm Child Psychol 2000; 28: 425–437.
Lahey BB, Applegate B, McBurnett K, Biederman J, Greenhill L, Hynd GW et al. DSM-IV field trials for attention deficit hyperactivity disorder in children and adolescents. Am J Psychiatry 1994; 151: 1673–1685.
Magnusson P, Smari J, Gretarsdottir H, Prandardottir H . Attention-deficit/hyperactivity symptoms in Icelandic schoolchildren: assessment with the attention deficit/hyperactivity rating scale-IV. Scand J Psychol 1999; 40: 301–306.
Baumgaertel A, Wolraich ML, Dietrich M . Comparison of diagnostic criteria for attention deficit disorders in a German elementary school sample. J Am Acad Child Adolesc Psychiatry 1995; 34: 629–638.
Rohde LA, Biederman J, Busnello EA, Zimmermann H, Schmitz M, Martins S et al. ADHD in a school sample of Brazilian adolescents: a study of prevalence, comorbid conditions, and impairments. J Am Acad Child Adolesc Psychiatry 1999; 38: 716–722.
Samuel VJ, George P, Thornell A, Curtis S, Taylor A, Brome D et al. A pilot controlled family study of DSM-III-R and DSM-IV ADHD in African-American children. J Am Acad Child Adolesc Psychiatry 1999; 38: 34–39.
Samuel VJ, Biederman J, Faraone SV, George P, Mick E, Thornell A et al. Clinical characteristics of attention deficit hyperactivity disorder in African American children. Am J Psychiatry 1998; 155: 696–698.
Taylor E, Sandberg S . Hyperactive behavior in English schoolchildren: a questionnaire survey. J Abnorm Child Psychol 1984; 12: 143–155.
Auerbach JG, Lerner Y . Syndromes derived from the child behavior checklist for clinically referred Israeli boys aged 6–11: a research note. J Child Psychol Psychiatry 1991; 32: 1017–1024.
Crijnen AA, Achenbach TM, Verhulst FC . Problems reported by parents of children in multiple cultures: the child behavior checklist syndrome constructs. Am J Psychiatry 1999; 156: 569–574.
Rasmussen ER, Todd RD, Neuman RJ, Heath AC, Reich W, Rohde LA . Comparison of male adolescent-report of attention-deficit/hyperactivity disorder (ADHD) symptoms across two cultures using latent class and principal components analysis. J Child Psychol Psychiatry 2002; 43: 797–805.
Weine AM, Phillips JS, Achenbach TM . Behavioral and emotional problems among Chinese and American children: parent and teacher reports for ages 6 to 13. J Abnorm Child Psychol 1995; 23: 619–639.
Rohde LA, Szobot C, Polanczyk G, Schmitz M, Martins S, Tramontina S . Attention-deficit/hyperactivity disorder in a diverse culture: do research and clinical findings support the notion of a cultural construct for the disorder? Biol Psychiatry 2005; 57: 1436–1441.
Lahey BB, Van Hulle CA, Singh AL, Waldman ID, Rathouz PJ . Higher-order genetic and environmental structure of prevalent forms of child and adolescent psychopathology. Arch Gen Psychiatry 2011; 68: 181–189.
Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 2010; 15: 1016–1022.
Rommelse NN, Franke B, Geurts HM, Hartman CA, Buitelaar JK . Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur Child Adolesc Psychiatry 2010; 19: 281–295.
Rommelse NN, Geurts HM, Franke B, Buitelaar JK, Hartman CA . A review on cognitive and brain endophenotypes that may be common in autism spectrum disorder and attention-deficit/hyperactivity disorder and facilitate the search for pleiotropic genes. Neurosci Biobehav Rev 2011; 35: 1363–1396.
Taurines R, Schmitt J, Renner T, Conner AC, Warnke A, Romanos M . Developmental comorbidity in attention-deficit/hyperactivity disorder. Atten Defic Hyperact Disord 2010; 2: 267–289.
Pliszka SR . Comorbidity of attention-deficit/hyperactivity disorder with psychiatric disorder: an overview. J Clin Psychiatry 1998; 59 (Suppl 7): 50–58.
Weber H, Kittel-Schneider S, Gessner A, Domschke K, Neuner M, Jacob CP et al. Cross-disorder analysis of bipolar risk genes: further evidence of DGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology 2011; 36: 2076–2085.
Reif A, Nguyen TT, Weissflog L, Jacob C, Romanos M, Renner T et al. DIRAS2 is associated with adult ADHD, related traits, and co-morbid disorders. Neuropsychopharmacology 2011; 36: 2318–2327.
Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Gottesman II, Gould TD . The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003; 160: 636–645.
Szatmari P, Maziade M, Zwaigenbaum L, Merette C, Roy MA, Joober R et al. Informative phenotypes for genetic studies of psychiatric disorders. Am J Med Genet B 2007; 144B: 581–588.
Kendler KS, Neale MC . Endophenotype: a conceptual analysis. Mol Psychiatry 2010; 15: 789–797.
Rommelse NN . Endophenotypes in the genetic research of ADHD over the last decade: have they lived up to their expectations? Expert Rev Neurother 2008; 8: 1425–1429.
Langley K, Heron J, O’Donovan MC, Owen MJ, Thapar A . Genotype link with extreme antisocial behavior: the contribution of cognitive pathways. Arch Gen Psychiatry 2010; 67: 1317–1323.
Castellanos FX, Tannock R . Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci 2002; 3: 617–628.
Nigg JT, Stavro G, Ettenhofer M, Hambrick DZ, Miller T, Henderson JM . Executive functions and ADHD in adults: evidence for selective effects on ADHD symptom domains. J Abnorm Psychol 2005; 114: 706–717.
Johnson KA, Wiersema JR, Kuntsi J . What would Karl Popper say? Are current psychological theories of ADHD falsifiable? Behav Brain Funct 2009; 5: 15.
Doyle AE, Faraone SV, Seidman LJ, Willcutt EG, Nigg JT, Waldman ID et al. Are endophenotypes based on measures of executive functions useful for molecular genetic studies of ADHD? J Child Psychol Psychiatry 2005; 46: 774–803.
Willcutt EG, Doyle AE, Nigg JT, Faraone SV, Pennington BF . Validity of the executive function theory of attention-deficit/hyperactivity disorder: a meta-analytic review. Biol Psychiatry 2005; 57: 1336–1346.
Pennington BF . Toward a new neuropsychological model of attention-deficit/hyperactivity disorder: subtypes and multiple deficits. Biol Psychiatry 2005; 57: 1221–1223.
Bidwell LC, Willcutt EG, DeFries JC, Pennington BF . Testing for neuropsychological endophenotypes in siblings discordant for attention-deficit/hyperactivity disorder. Biol Psychiatry 2007; 62: 991–998.
Rommelse NN, Altink ME, Oosterlaan J, Beem L, Buschgens CJ, Buitelaar J et al. Speed, variability, and timing of motor output in ADHD: which measures are useful for endophenotypic research? Behav Genet 2008; 38: 121–132.
Rommelse NN, Altink ME, Oosterlaan J, Buschgens CJ, Buitelaar J, Sergeant JA . Support for an independent familial segregation of executive and intelligence endophenotypes in ADHD families. Psychol Med 2008; 38: 1595–1606.
Slaats-Willemse D, Swaab-Barneveld H, de Sonneville L, van der ME, Buitelaar J . Deficient response inhibition as a cognitive endophenotype of ADHD. J Am Acad Child Adolesc Psychiatry 2003; 42: 1242–1248.
Crosbie J, Schachar R . Deficient inhibition as a marker for familial ADHD. Am J Psychiatry 2001; 158: 1884–1890.
Slaats-Willemse D, Swaab-Barneveld H, De SL, Buitelaar J . Familial clustering of executive functioning in affected sibling pair families with ADHD. J Am Acad Child Adolesc Psychiatry 2005; 44: 385–391.
Doyle AE, Willcutt EG, Seidman LJ, Biederman J, Chouinard VA, Silva J et al. Attention-deficit/hyperactivity disorder endophenotypes. Biol Psychiatry 2005; 57: 1324–1335.
Kuntsi J, Wood AC, Rijsdijk F, Johnson KA, Andreou P, Albrecht B et al. Separation of cognitive impairments in attention-deficit/hyperactivity disorder into 2 familial factors. Arch Gen Psychiatry 2010; 67: 1159–1167.
Halperin JM, Schulz KP . Revisiting the role of the prefrontal cortex in the pathophysiology of attention-deficit/hyperactivity disorder. Psychol Bull 2006; 132: 560–581.
Halperin JM, Trampush JW, Miller CJ, Marks DJ, Newcorn JH . Neuropsychological outcome in adolescents/young adults with childhood ADHD: profiles of persisters, remitters and controls. J Child Psychol Psychiatry 2008; 49: 958–966.
Gallagher R, Blader J . The diagnosis and neuropsychological assessment of adult attention deficit/hyperactivity disorder. Scientific study and practical guidelines. Ann N Y Acad Sci 2001; 931: 148–171.
Lijffijt M, Kenemans JL, Verbaten MN, van EH . A meta-analytic review of stopping performance in attention-deficit/hyperactivity disorder: deficient inhibitory motor control? J Abnorm Psychol 2005; 114: 216–222.
Malloy-Diniz L, Fuentes D, Leite WB, Correa H, Bechara A . Impulsive behavior in adults with attention deficit/ hyperactivity disorder: characterization of attentional, motor and cognitive impulsiveness. J Int Neuropsychol Soc 2007; 13: 693–698.
Antshel KM, Faraone SV, Maglione K, Doyle AE, Fried R, Seidman LJ et al. Executive functioning in high-IQ adults with ADHD. Psychol Med 2010; 40: 1909–1918.
Boonstra AM, Oosterlaan J, Sergeant JA, Buitelaar JK . Executive functioning in adult ADHD: a meta-analytic review. Psychol Med 2005; 35: 1097–1108.
Muller BW, Gimbel K, Keller-Pliessnig A, Sartory G, Gastpar M, Davids E . Neuropsychological assessment of adult patients with attention-deficit/hyperactivity disorder. Eur Arch Psychiatry Clin Neurosci 2007; 257: 112–119.
Schweitzer JB, Hanford RB, Medoff DR . Working memory deficits in adults with ADHD: is there evidence for subtype differences? Behav Brain Funct 2006; 2: 43.
King JA, Colla M, Brass M, Heuser I, von CD . Inefficient cognitive control in adult ADHD: evidence from trial-by-trial Stroop test and cued task switching performance. Behav Brain Funct 2007; 3: 42.
Boonstra AM, Kooij JJ, Oosterlaan J, Sergeant JA, Buitelaar JK . To act or not to act, that's the problem: primarily inhibition difficulties in adult ADHD. Neuropsychology 2010; 24: 209–221.
Bekker EM, Overtoom CC, Kenemans JL, Kooij JJ, De NI, Buitelaar JK et al. Stopping and changing in adults with ADHD. Psychol Med 2005; 35: 807–816.
Bekker EM, Overtoom CC, Kooij JJ, Buitelaar JK, Verbaten MN, Kenemans JL . Disentangling deficits in adults with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 2005; 62: 1129–1136.
Lampe K, Konrad K, Kroener S, Fast K, Kunert HJ, Herpertz SC . Neuropsychological and behavioural disinhibition in adult ADHD compared to borderline personality disorder. Psychol Med 2007; 37: 1717–1729.
Hervey AS, Epstein JN, Curry JF . Neuropsychology of adults with attention-deficit/hyperactivity disorder: a meta-analytic review. Neuropsychology 2004; 18: 485–503.
Kebir O, Tabbane K, Sengupta S, Joober R . Candidate genes and neuropsychological phenotypes in children with ADHD: review of association studies. J Psychiatry Neurosci 2009; 34: 88–101.
Boonstra AM, Kooij JJ, Buitelaar JK, Oosterlaan J, Sergeant JA, Heister JG et al. An exploratory study of the relationship between four candidate genes and neurocognitive performance in adult ADHD. Am J Med Genet B 2008; 147: 397–402.
Doyle AE, Ferreira MA, Sklar PB, Lasky-Su J, Petty C, Fusillo SJ et al. Multivariate genomewide linkage scan of neurocognitive traits and ADHD symptoms: suggestive linkage to 3q13. Am J Med Genet B 2008; 147B: 1399–1411.
Rommelse NN, Arias-Vasquez A, Altink ME, Buschgens CJ, Fliers E, Asherson P et al. Neuropsychological endophenotype approach to genome-wide linkage analysis identifies susceptibility loci for ADHD on 2q21.1 and 13q12.11. Am J Hum Genet 2008; 83: 99–105.
Aron AR, Gluck MA, Poldrack RA . Long-term test–retest reliability of functional MRI in a classification learning task. NeuroImage 2006; 29: 1000–1006.
Friedman L, Stern H, Brown GG, Mathalon DH, Turner J, Glover GH et al. Test–retest and between-site reliability in a multicenter fMRI study. Hum Brain Mapp 2008; 29: 958–972.
Noll DC, Genovese CR, Nystrom LE, Vazquez AL, Forman SD, Eddy WF et al. Estimating test–retest reliability in functional MR imaging. II: Application to motor and cognitive activation studies. Magn Reson Med 1997; 38: 508–517.
Fallgatter AJ, Bartsch AJ, Strik WK, Mueller TJ, Eisenack SS, Neuhauser B et al. Test–retest reliability of electrophysiological parameters related to cognitive motor control. Clin Neurophysiol 2001; 112: 198–204.
Mier D, Kirsch P, Meyer-Lindenberg A . Neural substrates of pleiotropic action of genetic variation in COMT: a meta-analysis. Mol Psychiatry 2010; 15: 918–927.
Munafo MR, Brown SM, Hariri AR . Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol Psychiatry 2008; 63: 852–857.
Valera EM, Faraone SV, Biederman J, Poldrack RA, Seidman LJ . Functional neuroanatomy of working memory in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 2005; 57: 439–447.
Valera EM, Brown A, Biederman J, Faraone SV, Makris N, Monuteaux MC et al. Sex differences in the functional neuroanatomy of working memory in adults with ADHD. Am J Psychiatry 2010; 167: 86–94.
Ehlis AC, Bahne CG, Jacob CP, Herrmann MJ, Fallgatter AJ . Reduced lateral prefrontal activation in adult patients with attention-deficit/hyperactivity disorder (ADHD) during a working memory task: a functional near-infrared spectroscopy (fNIRS) study. J Psychiatr Res 2008; 42: 1060–1067.
Wolf RC, Plichta MM, Sambataro F, Fallgatter AJ, Jacob C, Lesch KP et al. Regional brain activation changes and abnormal functional connectivity of the ventrolateral prefrontal cortex during working memory processing in adults with attention-deficit/hyperactivity disorder. Hum Brain Mapp 2009; 30: 2252–2266.
Karch S, Thalmeier T, Lutz J, Cerovecki A, Opgen-Rhein M, Hock B et al. Neural correlates (ERP/fMRI) of voluntary selection in adult ADHD patients. Eur Arch Psychiatry Clin Neurosci 2010; 260: 427–440.
Dibbets P, Evers L, Hurks P, Marchetta N, Jolles J . Differences in feedback- and inhibition-related neural activity in adult ADHD. Brain Cogn 2009; 70: 73–83.
Wiersema JR, van der Meere JJ, Roeyers H . ERP correlates of error monitoring in adult ADHD. J Neural Transm 2009; 116: 371–379.
Herrmann MJ, Mader K, Schreppel T, Jacob C, Heine M, Boreatti-Hummer A et al. Neural correlates of performance monitoring in adult patients with attention deficit hyperactivity disorder (ADHD). World J Biol Psychiatry 2010; 11: 457–464.
McLoughlin G, Albrecht B, Banaschewski T, Rothenberger A, Brandeis D, Asherson P et al. Performance monitoring is altered in adult ADHD: a familial event-related potential investigation. Neuropsychologia 2009; 47: 3134–3142.
Plichta MM, Vasic N, Wolf RC, Lesch KP, Brummer D, Jacob C et al. Neural hyporesponsiveness and hyperresponsiveness during immediate and delayed reward processing in adult attention-deficit/hyperactivity disorder. Biol Psychiatry 2009; 65: 7–14.
Strohle A, Stoy M, Wrase J, Schwarzer S, Schlagenhauf F, Huss M et al. Reward anticipation and outcomes in adult males with attention-deficit/hyperactivity disorder. NeuroImage 2008; 39: 966–972.
Cubillo A, Rubia K . Structural and functional brain imaging in adult attention-deficit/hyperactivity disorder. Expert Rev Neurother 2010; 10: 603–620.
Almeida LG, Ricardo-Garcell J, Prado H, Barajas L, Fernandez-Bouzas A, Avila D et al. Reduced right frontal cortical thickness in children, adolescents and adults with ADHD and its correlation to clinical variables: a cross-sectional study. J Psychiatr Res 2010; 44: 1214–1223.
Amico F, Stauber J, Koutsouleris N, Frodl T . Anterior cingulate cortex gray matter abnormalities in adults with attention deficit hyperactivity disorder: a voxel-based morphometry study. Psychiatry Res 2011; 191: 31–35.
Almeida Montes LG, Ricardo-Garcell J, Barajas De La Torre LB, Prado AH, Martinez Garcia RB, Fernandez-Bouzas A et al. Clinical correlations of grey matter reductions in the caudate nucleus of adults with attention deficit hyperactivity disorder. J Psychiatry Neurosci 2010; 35: 238–246.
Seidman LJ, Biederman J, Liang L, Valera EM, Monuteaux MC, Brown A et al. Gray matter alterations in adults with attention-deficit/hyperactivity disorder identified by voxel based morphometry. Biol Psychiatry 2011; 69: 857–866.
Frodl T, Stauber J, Schaaff N, Koutsouleris N, Scheuerecker J, Ewers M et al. Amygdala reduction in patients with ADHD compared with major depression and healthy volunteers. Acta Psychiatr Scand 2010; 121: 111–118.
Perlov E, Philipsen A, Tebartz van EL, Ebert D, Henning J, Maier S et al. Hippocampus and amygdala morphology in adults with attention-deficit hyperactivity disorder. J Psychiatry Neurosci 2008; 33: 509–515.
Makris N, Buka SL, Biederman J, Papadimitriou GM, Hodge SM, Valera EM et al. Attention and executive systems abnormalities in adults with childhood ADHD: a DT-MRI study of connections. Cereb Cortex 2008; 18: 1210–1220.
Mazaheri A, Coffey-Corina S, Mangun GR, Bekker EM, Berry AS, Corbett BA . Functional disconnection of frontal cortex and visual cortex in attention-deficit/hyperactivity disorder. Biol Psychiatry 2010; 67: 617–623.
Walitza S, Renner TJ, Dempfle A, Konrad K, Wewetzer C, Halbach A et al. Transmission disequilibrium of polymorphic variants in the tryptophan hydroxylase-2 gene in attention-deficit/hyperactivity disorder. Mol Psychiatry 2005; 10: 1126–1132.
Baehne CG, Ehlis AC, Plichta MM, Conzelmann A, Pauli P, Jacob C et al. Tph2 gene variants modulate response control processes in adult ADHD patients and healthy individuals. Mol Psychiatry 2009; 14: 1032–1039.
Dresler T, Ehlis AC, Heinzel S, Renner TJ, Reif A, Baehne CG et al. Dopamine transporter (SLC6A3) genotype impacts neurophysiological correlates of cognitive response control in an adult sample of patients with ADHD. Neuropsychopharmacology 2010; 35: 2193–2202.
van Dyck CH, Seibyl JP, Malison RT, Laruelle M, Zoghbi SS, Baldwin RM et al. Age-related decline in dopamine transporters: analysis of striatal subregions, nonlinear effects, and hemispheric asymmetries. Am J Geriatr Psychiatry 2002; 10: 36–43.
van de Giessen E, de Win MM, Tanck MW, van den Brink W, Baas F, Booij J . Striatal dopamine transporter availability associated with polymorphisms in the dopamine transporter gene SLC6A3. J Nucl Med 2009; 50: 45–52.
Brown AB, Biederman J, Valera EM, Doyle AE, Bush G, Spencer T et al. Effect of dopamine transporter gene (SLC6A3) variation on dorsal anterior cingulate function in attention-deficit/hyperactivity disorder. Am J Med Genet B 2010; 153B: 365–375.
Fallgatter AJ, Ehlis A-C, Dresler T, Reif A, Jacob CP, Lesch K-P . Influence of a Latrophilin 3 (LPHN3) risk haplotype on event-related potential measures of cognitive response control in attention-deficit/hyperactivity Disorder (ADHD), submitted.
Reif A, Jacob CP, Rujescu D, Herterich S, Lang S, Gutknecht L et al. Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch Gen Psychiatry 2009; 66: 41–50.
Hoogman M, Aarts E, Zwiers MP, Slaats-Willemse D, Naber M, Onnink M et al. Nitric Oxide Synthase genotype modulation of impulsivity and ventral striatal activity in attention deficit/hyperactivity disorder and control subjects. Am J Psychiatry 2011; 168: 1099–1106.
Loo SK, Hale ST, Hanada G, Macion J, Shrestha A, McGough JJ et al. Familial clustering and DRD4 effects on electroencephalogram measures in multiplex families with attention deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 368–377.
Monuteaux MC, Seidman LJ, Faraone SV, Makris N, Spencer T, Valera E et al. A preliminary study of dopamine D4 receptor genotype and structural brain alterations in adults with ADHD. Am J Med Genet B 2008; 147B: 1436–1441.
State MW . The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron 2010; 68: 254–269.
Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ et al. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. Am J Psychiatry 2011; 168: 930–946.
Ruano D, Abecasis GR, Glaser B, Lips ES, Cornelisse LN, de Jong AP et al. Functional gene group analysis reveals a role of synaptic heterotrimeric G proteins in cognitive ability. Am J Hum Genet 2010; 86: 113–125.
Liu J, Pearlson G, Windemuth A, Ruano G, Perrone-Bizzozero NI, Calhoun V . Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Hum Brain Mapp 2009; 30: 241–255.
Meda SA, Jagannathan K, Gelernter J, Calhoun VD, Liu J, Stevens MC et al. A pilot multivariate parallel ICA study to investigate differential linkage between neural networks and genetic profiles in schizophrenia. NeuroImage 2010; 53: 1007–1015.
Docherty SJ, Kovas Y, Petrill SA, Plomin R . Generalist genes analysis of DNA markers associated with mathematical ability and disability reveals shared influence across ages and abilities. BMC Genet 2010; 11: 61.
Schwender H, Ruczinski I, Ickstadt K . Testing SNPs and sets of SNPs for importance in association studies. Biostatistics 2011; 12: 18–32.
Ficks CA, Waldman ID . Gene–environment interactions in attention-deficit/hyperactivity disorder. Curr Psychiatry Rep 2009; 11: 387–392.
Pennington BF, McGrath LM, Rosenberg J, Barnard H, Smith SD, Willcutt EG et al. Gene × environment interactions in reading disability and attention-deficit/hyperactivity disorder. Dev Psychol 2009; 45: 77–89.
Wermter AK, Laucht M, Schimmelmann BG, Banaschweski T, Sonuga-Barke EJ, Rietschel M et al. From nature versus nurture, via nature and nurture, to gene × environment interaction in mental disorders. Eur Child Adolesc Psychiatry 2010; 19: 199–210.
Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, Sklar P et al. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 2009; 85: 13–24.
Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 2010; 15: 637–646.
Poelmans G, Pauls DL, Buitelaar JK, Franke B . Integrated genome-wide association study findings: identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am J Psychiatry 2011; 168: 365–377.
Morrison JR, Stewart MA . Bilateral inheritance as evidence for polygenicity in the hyperactive child syndrome. J Nerv Ment Dis 1974; 158: 226–228.
Faraone SV, Doyle AE . The nature and heritability of attention-deficit/hyperactivity disorder. Child Adolesc Psychiatr Clin N Am 2001; 10: 299-2ix.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al. Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Bassett AS, Scherer SW, Brzustowicz LM . Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am J Psychiatry 2010; 167: 899–914.
Merikangas AK, Corvin AP, Gallagher L . Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet 2009; 25: 536–544.
de Silva MG, Elliott K, Dahl HH, Fitzpatrick E, Wilcox S, Delatycki M et al. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J Med Genet 2003; 40: 733–740.
McKinney J, Johansson S, Halmoy A, Dramsdahl M, Winge I, Knappskog PM et al. A loss-of-function mutation in tryptophan hydroxylase 2 segregating with attention-deficit/hyperactivity disorder. Mol Psychiatry 2008; 13: 365–367.
Baker KL, Rees MI, Thompson PW, Howell RT, Cole TR, Houghes HE et al. Chromosome 2 interstitial deletion (del(2)(q14.1q21)) associated with connective tissue laxity and an attention deficit disorder. J Med Genet 2001; 38: 493–496.
Cappellacci S, Martinelli S, Rinaldi R, Martinelli E, Parisi P, Mancini B et al. De novo pure 12q22q24.33 duplication: first report of a case with mental retardation, ADHD, and Dandy–Walker malformation. Am J Med Genet A 2006; 140: 1203–1207.
Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 2010; 47: 332–341.
van den BL, de Waal HD, Han JC, Ylstra B, Eijk P, Nesterova M et al. Investigation of a patient with a partial trisomy 16q including the fat mass and obesity associated gene (FTO): fine mapping and FTO gene expression study. Am J Med Genet A 2010; 152A: 630–637.
Lalani SR, Thakuria JV, Cox GF, Wang X, Bi W, Bray MS et al. 20p12.3 microdeletion predisposes to Wolff–Parkinson–White syndrome with variable neurocognitive deficits. J Med Genet 2009; 46: 168–175.
Ingason A, Rujescu D, Cichon S, Sigurdsson E, Sigmundsson T, Pietilainen OP et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry 2011; 16: 17–25.
Lo-Castro A, D’Agati E, Curatolo P . ADHD and genetic syndromes. Brain Dev 2011; 33: 456–461.
Lopez-Arvizu C, Sparrow EP, Strube MJ, Slavin C, DeOleo C, James J et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in Nail-patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B 2011; 156B: 59–66.
Lesch KP, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry 2011; 16: 491–503.
Bradley WE, Raelson JV, Dubois DY, Godin E, Fournier H, Prive C et al. Hotspots of large rare deletions in the human genome. PLoS One 2010; 5: e9401.
Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009; 461: 272–276.
Shendure J, Ji H . Next-generation DNA sequencing. Nat Biotechnol 2008; 26: 1135–1145.
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB . Rare variants create synthetic genome-wide associations. PLoS Biol 2010; 8: e1000294.
Halmoy A, Johansson S, Winge I, McKinney JA, Knappskog P, Haavik J . ADHD symptoms in offspring of mothers with impaired serotonin production. Arch Gen Psychiatry 2010; 67: 1033–1043.
Hawi Z, Kent L, Hill M, Anney RJ, Brookes KJ, Barry E et al. ADHD and DAT1: further evidence of paternal over-transmission of risk alleles and haplotype. Am J Med Genet B 2010; 153B: 97–102.
Hawi Z, Segurado R, Conroy J, Sheehan K, Lowe N, Kirley A et al. Preferential transmission of paternal alleles at risk genes in attention-deficit/hyperactivity disorder. Am J Hum Genet 2005; 77: 958–965.
Hardy J . Genetic analysis of pathways to Parkinson disease. Neuron 2010; 68: 201–206.
Williams HJ, Craddock N, Russo G, Hamshere ML, Moskvina V, Dwyer S et al. Most genome-wide significant susceptibility loci for schizophrenia and bipolar disorder reported to date cross-traditional diagnostic boundaries. Hum Mol Genet 2011; 20: 387–391.
Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry 2009; 14: 252–260.
Froehlich TE, McGough JJ, Stein MA . Progress and promise of attention-deficit hyperactivity disorder pharmacogenetics. CNS Drugs 2010; 24: 99–117.
Kieling C, Genro JP, Hutz MH, Rohde LA . A current update on ADHD pharmacogenomics. Pharmacogenomics 2010; 11: 407–419.
Kooij SJ, Bejerot S, Blackwell A, Caci H, Casas-Brugue M, Carpentier PJ et al. European consensus statement on diagnosis and treatment of adult ADHD: The European Network Adult ADHD. BMC Psychiatry 2010; 10: 67.
Stein MA, McGough JJ . The pharmacogenomic era: promise for personalizing attention deficit hyperactivity disorder therapy. Child Adolesc Psychiatr Clin N Am 2008; 17: 475-xii.
Mick E, Neale B, Middleton FA, McGough JJ, Faraone SV . Genome-wide association study of response to methylphenidate in 187 children with attention-deficit/hyperactivity disorder. Am J Med Genet B 2008; 147B: 1412–1418.
Mick E, Biederman J, Spencer T, Faraone SV, Sklar P . Absence of association with DAT1 polymorphism and response to methylphenidate in a sample of adults with ADHD. Am J Med Genet B 2006; 141B: 890–894.
Kooij JS, Boonstra AM, Vermeulen SH, Heister AG, Burger H, Buitelaar JK et al. Response to methylphenidate in adults with ADHD is associated with a polymorphism in SLC6A3 (DAT1). Am J Med Genet B 2008; 147B: 201–208.
Contini V, Victor MM, Marques FZ, Bertuzzi GP, Salgado CA, Silva KL et al. Response to methylphenidate is not influenced by DAT1 polymorphisms in a sample of Brazilian adult patients with ADHD. J Neural Transm 2010; 117: 269–276.
Greven CU, Asherson P, Rijsdijk FV, Plomin R . A longitudinal twin study on the association between inattentive and hyperactive-impulsive ADHD symptoms. J Abnorm Child Psychol 2011; 39: 623–632.
Langley K, Fowler TA, Grady DL, Moyzis RK, Holmans PA, van den Bree MB et al. Molecular genetic contribution to the developmental course of attention-deficit hyperactivity disorder. Eur Child Adolesc Psychiatry 2009; 18: 26–32.
Huang J, Perlis RH, Lee PH, Rush AJ, Fava M, Sachs GS et al. Cross-disorder genomewide analysis of schizophrenia, bipolar disorder, and depression. Am J Psychiatry 2010; 167: 1254–1263.
Acknowledgements
The work of KPL and AR was supported by the Deutsche Forschungsgemeinschaft (KFO 125, SFB 581, SFB TRR 58, GRK 1156, GRK 1253) and the Bundesministerium für Bildung und Forschung (KPL, BMBF 01GV0605). BC is supported by the Agència de Gestió d’Ajuts Universitaris i de Recerca-AGAUR (2009GR00971) and JAR-Q by Health Department (Government of Catalonia), Alicia Koplowitz Foundation, Fundació La Marató de TV3 (092330/31) and Instituto de Salud Carlos III-FIS (PI080519). BF and JB are supported by the Netherlands Organization for Scientific Research (NWO, Brain and Cognition 433-09-229 and 433-09-242). CHDB is supported by the Brazilian funding agencies CNPq, CAPES, PRONEX and FAPERGS-DECIT-PPSUS. JH and SJ receive support from the Research Council of Norway, the KG Jebsen Centre for Research on Neuropsychiatric Disorders and the Western Norway Regional Health Authority.
Author information
Authors and Affiliations
Consortia
Corresponding author
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Franke, B., Faraone, S., Asherson, P. et al. The genetics of attention deficit/hyperactivity disorder in adults, a review. Mol Psychiatry 17, 960–987 (2012). https://doi.org/10.1038/mp.2011.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2011.138
Keywords
This article is cited by
-
Genome-wide Mendelian randomization identifies actionable novel drug targets for psychiatric disorders
Neuropsychopharmacology (2023)
-
Gray matter volumetric correlates of attention deficit and hyperactivity traits in emerging adolescents
Scientific Reports (2022)
-
Molecular Characterisation of the Mechanism of Action of Stimulant Drugs Lisdexamfetamine and Methylphenidate on ADHD Neurobiology: A Review
Neurology and Therapy (2022)
-
Systematic review of mitochondrial genetic variation in attention-deficit/hyperactivity disorder
European Child & Adolescent Psychiatry (2022)
-
Kohortenstudien in der Kinder- und Jugendpsychiatrie
Der Nervenarzt (2021)
