
Abstract
To evaluate the diagnostic value of MDM2 status in craniofacial fibro-osseous lesions, we investigated MDM2 expression by immunohistochemistry and analyzed MDM2 amplification by qPCR in 30 cases of ossifying fibroma (including 13 cases of the juvenile variant) and 17 cases of fibrous dysplasia. Two cases of uncommon extragnathic psammomatoid fibrous dysplasia and a mixed control group of 15 cases of low-grade osteosarcoma and 15 cases of well-differentiated/dedifferentiated liposarcoma were included. MDM2 amplification was found in 33% of ossifying fibromas (peak of 69% for the juvenile variant) and in 12% of fibrous dysplasia, in none of which was MDM2 overexpressed. All control cases exhibited MDM2 amplification and overexpression. To investigate possible polysomy of chromosome 12, we studied RASAL1 amplification, a gene telomeric to MDM2 on the long arm of chromosome 12. RASAL1 amplification was reported in all benign fibro-osseous lesions exhibiting MDM2 amplification but not in controls. Simultaneous amplification of these two genes was significantly higher in juvenile ossifying fibromas compared with fibrous dysplasia (P=0.004), non-juvenile ossifying fibromas (P=0.001), and all other benign craniofacial fibro-osseous lesions combined (P=0.0001). Of the nine cases of juvenile ossifying fibroma exhibiting amplification, three were locally invasive and four were recurrent, suggesting aggressive disease. The two cases of extragnathic psammomatoid fibrous dysplasia also showed MDM2 and RASAL1 amplification with no MDM2 overexpression. This large chromosome 12 rearrangement, spanning MDM2 and RASAL1, is the first recurrent molecular abnormality to be reported in juvenile ossifying fibroma. It may represent both a molecular diagnostic marker and a characteristic of more aggressive forms with a higher risk of recurrence. Finally, the presence of this rearrangement in extragnathic psammomatoid fibro-osseous lesions mimicking ossifying fibromas might reflect a common molecular pathway in their pathogenesis and calls into question the classification of such lesions within fibrous dysplasia.
Similar content being viewed by others


Main
Diagnosis of fibro-osseous lesions requires a combination of clinical, radiological, histological, and molecular findings. The most frequent fibro-osseous lesions in the craniofacial region are fibrous dysplasia and ossifying fibroma (the latter being reported only in the craniofacial area) and more rarely low-grade osteosarcoma.1 Although these lesions frequently overlap in morphology, it is important to distinguish between them because their evolution, prognosis, and management differ.2, 3, 4 Morphologically, the lesions share a mixed fibrous component comprising varying proportions of small ovoid or spindle cells, associated with a variably mature bone component consisting of spherules and/or trabeculae of woven bone. In the craniofacial area, particular morphological features can confuse the diagnosis. The osteogenesis of fibrous dysplasia varies from thick anastomosing trabeculae, a pattern reminiscent of low-grade osteosarcoma, to small calcified spherules that mimic cement and may be barely distinguishable from ossifying fibroma.5, 6, 7 In addition to its conventional form, the World Health Organization (WHO) recognizes two clinicopathological variants of ossifying fibroma, ie, psammomatoid juvenile ossifying fibroma and trabecular juvenile ossifying fibroma.1 These lesions develop in any facial bones, mostly in individuals around the age of 15 years, and most frequently in the maxilla and the mandible.8 Juvenile ossifying fibroma is a distinct clinicopathological entity among fibro-osseous lesions because of its rapid growth, its potential for local invasiveness and its tendency to recur (20–90%).9, 10 Surgical excision of juvenile ossifying fibroma is thus considered necessary.11 Immunohistochemical and molecular markers have been developed to assist diagnosis of these challenging fibro-osseous lesions. Distinguishing fibrous dysplasia and low-grade osteosarcoma from other fibro-osseous lesions is currently based on the presence of GNAS (guanine nucleotide-binding protein/alpha subunit) mutations and MDM2 (murine double-minute 2) amplification/overexpression, respectively. The pathogenesis of fibrous dysplasia is partially explained by GNAS mutations, which induce constitutive activation of a G-protein alpha subunit.12, 13 These mutations have not been described in other fibro-osseous lesions and roughly 50% of fibrous dysplasia lesions are found to be mutated.14, 15 Recent studies have not identified GNAS mutations in ossifying fibroma.15, 16, 17, 18
Cytogenetic abnormalities such as supernumerary ring chromosomes, including amplification of chromosome 12q13-15 leading to multiple copies of MDM2, characterize low-grade osteosarcoma.19, 20, 21 To differentiate low-grade osteosarcoma from other fibro-osseous lesions, several studies have shown the diagnostic value of detecting overexpression of MDM2 by immunohistochemistry,22, 23 and MDM2 amplification by quantitative real-time PCR (qPCR), comparative genomic hybridization (CGH) array, or fluorescence in situ hybridization (FISH).22 These studies mostly included cases of fibrous dysplasia and only a few cases of ossifying fibroma (six cases in Dujardin et al and none in Yoshida et al). No studies have specifically evaluated MDM2 status in craniofacial fibro-osseous lesions to date.
The aim of this study was to evaluate the diagnostic value of MDM2 overexpression and gene amplification in craniofacial fibro-osseous lesions. We studied 47 cases of benign craniofacial fibro-osseous lesions, comprising 30 cases of ossifying fibroma and 17 cases of fibrous dysplasia. Because of morphological similarity to the psammomatoid juvenile variant of ossifying fibroma, we included two extragnathic cases of the uncommon psammomatoid subtype of fibrous dysplasia. Fifteen cases of low-grade osteosarcoma (including one jawbone location) and 15 cases of well-differentiated/dedifferentiated liposarcoma, known to exhibit MDM2 overexpression and amplification, were studied as controls.
Materials and methods
Selection of Patients and Samples and DNA Extraction
This retrospective study was designed in conformance with our institutional ethics guidelines. Between 2000 and 2013, 47 cases of benign craniofacial fibro-osseous lesions were diagnosed morphologically at the University Hospital of Tours (France), including 30 cases of ossifying fibroma and 17 cases of fibrous dysplasia. We selected representative tissues (formalin-fixed and paraffin-embedded (FFPE)). Hematoxylin-eosin-and-saffron slides were reviewed by two pathologists (GdP and FTD) before immunostaining and molecular studies.
Genomic DNA was extracted using the QIAamp DNA FFPE Extraction kit (Qiagen, Courtaboeuf, France) from 10 μm sections cut from tissue blocks from 41 cases and snap-frozen tissue samples (stored at −80 °C) from six cases were extracted using the QIAamp DNA Mini Kit (Qiagen). Thick sections of FFPE tissue were dewaxed by extraction in 100% xylene and washed with 100% ethanol. Samples were air-dried before DNA extraction. Extracted DNA was quantified using a spectrophotometer (Cary100Scan Varian, Agilent, Massy, France). We included two uncommon cases of psammomatoid fibrous dysplasia, one located in the tibia and one in the talus, morphologically mimicking ossifying fibroma of the jaw due to the presence of small osteoid globules as cementiform particles. As controls for MDM2 overexpression and amplification, we studied 15 cases of low-grade osteosarcoma, including one occurring in the jawbone (13 from FFPE and 2 from frozen samples) and 15 cases of well-differentiated/dedifferentiated liposarcoma (14 from frozen samples and 1 from FFPE) known to exhibit MDM2 overexpression and amplification.
MDM2 Immunohistochemistry
For MDM2 immunostaining, 4-μm-thick paraffin sections were cut and mounted on glass slides (Superfrost, Dako, Trappes, France). Preparations were dried for 1 h at 58 °C, then overnight at 37 °C, sections were deparaffinized with toluene and rehydrated in ethanol. They were then pretreated with EDTA (MS-Unmasker-Diapath, Martinengo, Italy) and a heat-based antigen retrieval method was used before incubation. Endogenous peroxidase was blocked using 3% H2O2 for 5 min. The primary MDM2 antibody was used at 1:25 dilution (Zymed Laboratories, CA, USA; clone IF2) and sections were incubated for 1 h at 22 °C. Staining was performed with the Envision kit (Dako, Carpinteria, CA, USA); sections were revealed in a diaminobenzidine solution for 7 min and stained with hematoxylin for 16 s. MDM2 immunostaining was performed for each case of ossifying fibroma, fibrous dysplasia, low-grade osteosarcoma, and well-differentiated/dedifferentiated liposarcoma. Immunoreactivity was considered as negative when no cells or <1% of cells were stained and was considered as positive when 1–100% of cells were stained, irrespective of the intensity of staining. Only well-defined nuclear reactivity was considered positive.
Molecular Studies
qPCR for MDM2
qPCR for MDM2 amplification was performed on each case of ossifying fibroma and fibrous dysplasia (47 cases) and on each case of low-grade osteosarcoma and well-differentiated/dedifferentiated liposarcoma (30 cases). qPCR was performed using a LightCycler 480 (Roche, Boulogne, France) with 50 ng DNA using the Sybr Green I Master kit (Roche). The ALB gene was used as a reference. Primer sequences are given in Table 1. PCR was carried out as follows: following 5 min at 95 °C, 45 amplification cycles were run, each consisting of 10 s at 95 °C, 10 s at 60 °C and 10 s at 72 °C. The relative amount of MDM2 was established as a ratio of the reference gene (ALB) using LightCycler Relative Quantification software (Roche). We considered that all ratios above 2.5 indicated MDM2 amplification.
qPCR for RASAL1, IPO8, and KLRG1
To challenge the hypothesis of chromosome 12 polysomy for cases exhibiting MDM2 amplification, we studied amplification of one gene on the long arm of chromosome 12, ie, RASAL1 (RAS protein activator like-1), telomeric to MDM2 (12 cases), and two genes on the short arm of chromosome 12 (6 cases), ie, IPO8 (importin 8) and KLRG1 (killer cell lectin-like receptor subfamily G member 1). qPCR was performed using the same protocol as described for MDM2, with ALB as reference. All ratios above 2.5 were indicative of amplification of RASAL1, IPO8, or KLRG1. Primer sequences are given in Table 1.
Statistical Analysis
A Pearson χ2-test was performed to determine whether the presence of MDM2 amplification was significantly linked to the diagnosis of any craniofacial fibro-osseous lesions. XLStat software was used for statistical analysis (Addinsoft, Paris, France). P-values <0.05 were considered significant.
Results
Clinical and Pathology Findings
Ossifying fibroma
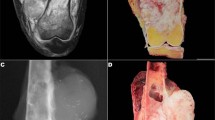
We report clinical and pathological findings for the 30 patients with ossifying fibroma. All had a suitable quantity and/or quality of extracted DNA for molecular analysis for MDM2 amplification (Table 2). Ages ranged from 1 to 68 years (mean 25 years). The male-to-female ratio was 0.6 (male 12; female 18). The most frequent site was the mandible (nine cases), followed by the sinuses (eight cases; maxilla, frontal, sphenoid, or ethmoid), the maxilla (seven cases), the orbit (one case), and the parietal skull bone (one case). Thirteen patients had a juvenile form of ossifying fibroma, 10 of which were trabecular and 3 were psammomatoid variants. The mean age of these patients was 15 years (range 8–24 years) and the sex ratio was 0.9 (6 males; 7 females). In this group, five had relapsed between 10 months and 21 years after surgery, whereas no recurrence occurred in other cases of ossifying fibroma. The juvenile form was associated with rapid growth of the tumor mass in five cases. Figure 1 illustrates representative histological features for the two forms of juvenile ossifying fibroma. The trabecular variant showed zonal architecture, with dense central cellular areas and low numbers of cells in peripheral areas (Figure 1a). In cellular fibroblastic areas, osteogenesis appeared immature and was often limited to densification of the collagen network. The bone trabeculae gradually matured and were lined by osteoblasts, giving the appearance of osteoblastoma (Figure 1b). The psammomatoid variant of juvenile ossifying fibroma showed a cellular background of plump fibroblasts associated with numerous small calcified cementicles (Figure 1c).

Histological appearance of juvenile trabecular variant of ossifying fibroma with immature osteogenesis (a) or an ‘osteoblastoma-like’ pattern (b), juvenile psammomatoid ossifying fibroma (c), cementifying form of extra-craniofacial fibrous dysplasia, mimicking juvenile psammomatoid variant of ossifying fibroma (d).
Craniofacial fibrous dysplasia
The mean age of the 17 patients with craniofacial fibrous dysplasia was 25 years (10–49 years) and the sex ratio was 0.7 (7 males; 10 females). Tumor sites were the sinus (five patients), the maxilla (five cases), the skull (four cases; parietal, temporal or frontal), the orbit (two cases), and the mandible (one case). Histologically, 11 patients had conventional fibrous dysplasia and 6 had unconventional fibrous dysplasia: 4 osteosclerosing, 1 fibro-involutive, and 1 with prominent aneurysmal cystic changes, as previously described.15
Psammomatoid extra-craniofacial fibrous dysplasia
We also studied two unusual cases of extra-craniofacial psammomatoid fibrous dysplasia because of their morphological similarity to ossifying fibroma. One was a 26-year-old woman with an osteolytic lesion of the tibia and the other a 60-year-old woman with a lesion of the talus. These fibro-osseous lesions were morphologically similar to ossifying fibroma, with abundant osteogenesis exclusively composed of small osteoid globules like cementicles and a cellular background, as observed in juvenile variants of ossifying fibroma (Figure 1d).
Low-grade osteosarcoma and well-differentiated/dedifferentiated liposarcoma (control group)
A group of 15 cases of low-grade osteosarcoma and 15 of well-differentiated/dedifferentiated liposarcoma (11 and 4 cases, respectively) were studied as controls for amplification. The most frequent site of the osteosarcomas was the femur (eight cases). One 22-year-old girl had an osteosarcoma located on the mandible. The most frequent site for liposarcoma was the lower limb (eight cases).
Immunohistochemistry
MDM2 overexpression was not observed in any of the 47 cases of benign craniofacial fibro-osseous lesions. In the control group, 10 out of 15 low-grade osteosarcomas and all the well-differentiated/dedifferentiated liposarcomas showed overexpression of MDM2.
Molecular Findings
qPCR of MDM2
Twelve of the 47 (26%) benign craniofacial fibro-osseous lesions showed MDM2 amplification by qPCR. This included 10 of the 30 (33%) cases of ossifying fibroma. Interestingly, MDM2 amplification was seen in 9 of the 13 (69%) juvenile variants (3-fold to 13-fold relative amplification), 2 of the 3 cases of psammomatoid variants and 7 of the 10 cases of trabecular variants. Amplification was reported in 2 of the 17 (12%) cases of craniofacial fibrous dysplasia (fivefold and eightfold relative amplification). In the control group of 13 cases of low-grade osteosarcoma with available DNA, all exhibited MDM2 amplification (3-fold to 21-fold relative amplification). In particular, the five cases with no immunohistochemical MDM2 overexpression had MDM2 amplification. In the group of well-differentiated/dedifferentiated liposarcomas, all cases exhibited MDM2 amplification (3-fold to 40-fold relative amplification) with MDM2 overexpression.
qPCR of RASAL1, IPO8, and KLRG1
Given the absence of MDM2 overexpression, we hypothesized that MDM2 amplification might have a particular molecular form involving a large chromosomal rearrangement or complete polysomy of chromosome 12. To evaluate this hypothesis, we investigated amplification levels of other genes, one located on the long arm of chromosome 12, telomeric to MDM2, ie, RASAL1, and two located on the short arm of chromosome 12, ie, IPO8 and KLRG1. None of these genes are known to trigger osteogenic transformation or to be amplified in cancerous lesions. Interestingly, in all fibro-osseous lesions exhibiting MDM2 amplification, we observed amplification of RASAL1 (3-fold to 20-fold relative amplification in ossifying fibroma and 3-fold to 10-fold in fibrous dysplasia), whereas RASAL1 amplification was not detected in the control groups of low-grade osteosarcoma or well-differentiated/dedifferentiated liposarcomas.
The simultaneous amplification of MDM2 and RASAL1 was significantly more frequent in the group of juvenile ossifying fibromas compared with fibrous dysplasia (P=0.004), with all other benign craniofacial lesions (fibrous dysplasia and non-juvenile ossifying fibroma, 69% vs 9%, P=0.0001), and with non-juvenile ossifying fibroma (69% vs 6%, P=0.001). P-values are summarized in Table 3. None of the six cases of ossifying fibroma investigated, amplified for MDM2 and RASAL1, showed amplification of IPO8 or KLRG1 (not shown).
The two cases of psammomatoid fibrous dysplasia mimicking juvenile psammomatoid ossifying fibroma also showed MDM2 and RASAL1 amplification (6-fold and 40-fold relative amplification for MDM2 and 5-fold and 21-fold for RASAL1).
Discussion
Craniofacial fibro-osseous lesions represent a diagnostic challenge because of overlaps and similarities in their clinical, radiological, and morphological characteristics. Investigating GNAS mutations in these lesions is appropriate to rule out fibrous dysplasia. However, routine GNAS genotyping has proven to be difficult on formalin-fixed, decalcified, and paraffin-embedded bone tissues,15 and no immunohistochemical marker for GNAS mutation is currently available.24
Although rare in jawbone locations,25, 26, 27 diagnosis of low-grade osteosarcoma should be considered in the context of fibro-osseous lesions due to its specific management.28, 29 Overexpression and gene amplification of MDM2 in low-grade/dedifferentiated osteosarcomas, linked to supernumerary ring 12q13-15 chromosomes containing MDM2,22, 30 have together been proven to be a sensitive and specific diagnostic tool for these tumors in extragnathic locations. Unlike fibrous dysplasia and low-grade osteosarcoma, no recurrent genetic abnormalities have been reported for ossifying fibroma, which remains a lesion of uncertain nature, arising from the periodontal ligament.31 Isolated cytogenetic abnormalities have been reported in rare cases but no recurrent genetic modification.32, 33, 34, 35 Zhang et al recently suggested that modified Notch signaling (Notch being involved in the control of cell differentiation and proliferation in normal bone) might be a molecular marker of ossifying fibroma.36
The juvenile variants of ossifying fibroma include two clinicopathological entities, ie, trabecular and psammomatoid juvenile ossifying fibroma. There are differences between these two juvenile variants including the site and age at occurrence. Psammomatoid juvenile ossifying fibroma predominantly affects the sinonasal and orbital bones of the skull, whereas trabecular juvenile ossifying fibroma occurs mostly in the jaw, with a predilection for the maxilla. The psammomatoid variant affects patients who are older (average age range 16–33 years) than the trabecular variant (8–12 years), and a wider range of ages (3 months to 72 years compared with 2–12 years for the trabecular variant) (for review see El-Mofty et al37).
From a morphological point of view, the two juvenile variants differ according to osteogenesis, with small uniform spherical ossicles for the psammomatoid variant and trabeculae of woven bone for the trabecular variant. The two variants share the same dense fibroblastic fibrous background and plump osteoblasts lining the osseous component, giving in some cases an osteoblastoma-like appearance. This explains why Makek first used the term psammous desmo-osteoblastoma and trabecular desmo-osteoblastoma to describe these two types of juvenile ossifying fibroma.38, 39 Despite these clinicopathological differences, both juvenile variants share a potentially expansive and locally aggressive evolution in younger patients.1, 11, 40 The prognosis, and therefore the clinical management, of these lesions remain uncertain.41 No clinical or radiological parameters, and no specific morphological findings or biomarkers can currently predict the aggressive evolution or risk of recurrence of juvenile ossifying fibroma.42, 43
In this study, we evaluated the diagnostic value of overexpression and gene amplification of MDM2 in a cohort of patient with benign craniofacial fibro-osseous lesions. We found MDM2 amplification in 26% of the cases, with a peak at 69% for juvenile ossifying fibroma. To our knowledge, no recurrent modification involving MDM2 in benign fibro-osseous lesions has been described to date. Unlike what was found in low-grade osteosarcomas and well-differentiated/dedifferentiated liposarcomas, MDM2 overexpression was not observed in benign craniofacial fibro-osseous lesions with MDM2 amplification.
Our aim was to characterize the chromosomal rearrangement containing the amplified MDM2 locus by evaluating RASAL1 amplification, another gene located on the long arm of chromosome 12, and the amplification of IPO8 and KLRG1 on the short arm of chromosome 12. RASAL1 amplification was found in all cases of benign craniofacial fibro-osseous lesions exhibiting MDM2 amplification, whereas no RASAL1 amplification was observed in the control group of low-grade osteosarcomas and well-differentiated/dedifferentiated liposarcomas, suggesting a different mechanism of amplification. This simultaneous amplification of two distant genes on the long arm of chromosome 12 is suggestive of a large rearrangement of this chromosome. Such a rearrangement might be more conservative of the chromatin structure and could explain why MDM2 amplification was not associated with MDM2 overexpression. The precise extent of the genetic modification on chromosome 12 in benign craniofacial fibro-osseous lesions remains to be characterized. A larger cohort studied with CGH array or next-generation sequencing (NGS) should help to define this chromosomal abnormality.
Several hypotheses can be formulated to explain this genetic modification. Polysomy of chromosome 12, as observed in spindle cell lipomas,44 is possible but would be unusual if involving only the long arm of chromosome 12, given the absence of amplification of IPO8 and KLRG1. Furthermore, it would be difficult to explain the very high amplification levels observed in several lesions (up to 20-fold relative amplification in juvenile ossifying fibroma) by the presence of a partial isochromosome 12. One alternative mechanism might be the presence of a supplementary ring chromosome including a repetition of a large locus containing MDM2 and RASAL1. Unfortunately, the FISH and CGH array studies attempted on two and six cases, respectively, of ossifying fibroma with MDM2 and RASAL1 amplification using FFPE tissue were technically inconclusive (data not shown). This might be explained by the stringent decalcification of the samples, leading to poor quality samples.
Although not clearly characterized, the chromosome 12 rearrangement involving MDM2 and RASAL1 is the first recurrent molecular abnormality reported in benign craniofacial fibro-osseous lesions, and more specifically in juvenile ossifying fibromas. Given that the amplification was found in 69% of juvenile ossifying fibromas it may well constitute a useful molecular tool for diagnosing juvenile ossifying fibroma. Moreover, among the nine cases of juvenile ossifying fibroma with MDM2 and RASAL1 amplification, three demonstrated local invasiveness or rapid growth, and four were recurrent. Thus, MDM2 and RASAL1 amplification appears indicative of aggressive lesions. On the other hand, this amplification was associated with a less carcinogenic phenotype compared with the isolated amplification of MDM2 in osteosarcoma.21 This difference might relate to RASAL1 amplification, which is known to counteract transformation in colorectal cancer.45 RASAL2, a closely related member in the RASAL1 family, has also been shown to suppress tumorigenicity and metastasis.46 This general characteristic of RASAL genes needs further investigation to obtain confirmation.
Interestingly, the two cases of psammomatoid fibrous dysplasia of the talus and the tibia exhibited the MDM2 and RASAL1 amplification. One of these lesions relapsed, emphasizing the aggressive and recurrent nature of lesions showing amplification of the 12q region covering MDM2 and RASAL1. Moreover, the absence of GNAS mutations in these two cases (data not shown) does not completely exclude the diagnosis of psammomatoid fibrous dysplasia, but raises the question of whether psammomatoid fibro-osseous lesions of the appendicular skeleton represent a variant of fibrous dysplasia or a distinct entity. Ossifying fibromas are currently only individualized as lesions of the craniofacial bones.47 Some extra-craniofacial lesions with trabecular pattern, previously described as ossifying fibroma, are now considered to be osteofibrous dysplasia in the tibia.48 The extra-craniofacial lesions morphologically similar to juvenile psammomatoid ossifying fibroma remain difficult to classify. In a cytogenetic study of fibro-osseous lesions of bone in children, Parham et al reported karyotypic features for a case of fibro-osseous lesion of the talus in a 7-year-old girl, mimicking juvenile psammomatoid ossifying fibroma of the jaw and for three cases of ossifying fibroma of the orbit in adolescents (13–17 years of age). The tumors had complex karyotypes with multiple translocations, but with common break points on Xq26 and 2q33.49 These cytogenetic abnormalities do not occur in fibrous dysplasia. The histological and molecular features of our two cases of psammomatoid fibrous dysplasia and the histological and cytogenetic findings of the few cases reported by Parham et al strongly suggest the existence of an extragnathic ‘ossifying fibroma’ entity.
In conclusion, modified chromosome 12 harboring a broad amplification encompassing MDM2 and RASAL1 is the first recurrent molecular abnormality reported to characterize juvenile ossifying fibromas. This abnormality is not only a potential molecular diagnostic marker, but may also be indicative of aggressive forms of the disease, with a higher risk of recurrence. The psammomatoid type of fibrous dysplasia occurring at extra-craniofacial sites appeared molecularly close to a psammomatoid variant of juvenile ossifying fibroma, in sharing MDM2 and RASAL1 amplification. Molecular studies using non-decalcified tissues or other extraction procedures will, by allowing better preservation of DNA integrity, make possible precise cartography of the rearrangement through CGH array or NGS, and will ultimately improve our understanding of the etiological molecular pathway involved.
References
Barnes L, Eveson JW, Reichart P et al. Pathology and Genetics of Head and Neck Tumours. IARC Press: Lyon, 2005, p 430.
Brannon RB, Fowler CB . Benign fibro-osseous lesions: a review of current concepts. Adv Anat Pathol 2001;8:126–143.
Alawi F . Benign fibro-osseous diseases of the maxillofacial bones. A review and differential diagnosis. Am J Clin Pathol 2002;118 (Suppl):S50–S70.
Suarez-Soto A, Baquero-Ruiz de la Hermosa MC, Minguez-Martinez I et al. Management of fibro-osseous lesions of the craniofacial area. Presentation of 19 cases and review of the literature. Med Oral Patol Oral Cir Bucal 2013;18:e479–e485.
Voytek TM, Ro JY, Edeiken J et al. Fibrous dysplasia and cemento-ossifying fibroma. A histologic spectrum. Am J Surg Pathol 1995;19:775–781.
Riminucci M, Liu B, Corsi A et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol 1999;187:249–258.
Bertoni F, Bacchini P, Fabbri N et al. Osteosarcoma. Low-grade intraosseous-type osteosarcoma, histologically resembling parosteal osteosarcoma, fibrous dysplasia, and desmoplastic fibroma. Cancer 1993;71:338–345.
Slootweg PJ, Panders AK, Koopmans R et al. Juvenile ossifying fibroma. An analysis of 33 cases with emphasis on histopathological aspects. J Oral Pathol Med 1994;23:385–388.
Johnson LC, Yousefi M, Vinh TN et al. Juvenile active ossifying fibroma. Its nature, dynamics and origin. Acta Otolaryngol Suppl 1991;488:1–40.
Slootweg PJ . Juvenile trabecular ossifying fibroma: an update. Virchows Arch 2012;461:699–703.
Sarode SC, Sarode GS, Waknis P et al. Juvenile psammomatoid ossifying fibroma: a review. Oral Oncol 2011;47:1110–1116.
Alman BA, Greel DA, Wolfe HJ . Activating mutations of Gs protein in monostotic fibrous lesions of bone. J Orthop Res 1996;14:311–315.
Marie P . Cellular and molecular biology of fibrous dysplasia. Ann Pathol 2001;21:489–498.
Lee SE, Lee EH, Park H et al. The diagnostic utility of the GNAS mutation in patients with fibrous dysplasia: meta-analysis of 168 sporadic cases. Hum Pathol 2012;43:1234–1242.
Tabareau-Delalande F, Collin C, Gomez-Brouchet A et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions: a retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol 2013;26:911–921.
Toyosawa S, Yuki M, Kishino M et al. Ossifying fibroma vs fibrous dysplasia of the jaw: molecular and immunological characterization. Mod Pathol 2007;20:389–396.
Patel MM, Wilkey JF, Abdelsayed R et al. Analysis of GNAS mutations in cemento-ossifying fibromas and cemento-osseous dysplasias of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:739–743.
Shi RR, Li XF, Zhang R et al. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol 2013;26:1023–1031.
Szymanska J, Mandahl N, Mertens F et al. Ring chromosomes in parosteal osteosarcoma contain sequences from 12q13-15: a combined cytogenetic and comparative genomic hybridization study. Genes Chromosomes Cancer 1996;16:31–34.
Tarkkanen M, Bohling T, Gamberi G et al. Comparative genomic hybridization of low-grade central osteosarcoma. Mod Pathol 1998;11:421–426.
Park HR, Jung WW, Bertoni F et al. Molecular analysis of p53, MDM2 and H-ras genes in low-grade central osteosarcoma. Pathol Res Pract 2004;200:439–445.
Dujardin F, Binh MB, Bouvier C et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol 2011;24:624–637.
Yoshida A, Ushiku T, Motoi T et al. Immunohistochemical analysis of MDM2 and CDK4 distinguishes low-grade osteosarcoma from benign mimics. Mod Pathol 2010;23:1279–1288.
Furukawa T, Kuboki Y, Tanji E et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 2011;1:161.
Jasnau S, Meyer U, Potratz J et al. Craniofacial osteosarcoma experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol 2008;44:286–294.
Amaral MB, Buchholz I, Freire-Maia B et al. Advanced osteosarcoma of the maxilla: a case report. Med Oral Patol Oral Cir Bucal 2008;13:492–495.
Thariat J, Julieron M, Brouchet A et al. Osteosarcomas of the mandible: are they different from other tumor sites? Crit Rev Oncol Hematol 2012;82:280–295.
Nthumba PM . Osteosarcoma of the jaws: a review of literature and a case report on synchronous multicentric osteosarcomas. World J Surg Oncol 2012;10:240.
Thariat J, Schouman T, Brouchet A et al. Osteosarcomas of the mandible: multidisciplinary management of a rare tumor of the young adult a cooperative study of the GSF-GETO, Rare Cancer Network, GETTEC/REFCOR and SFCE. Ann Oncol 2013;24:824–831.
Yoshida A, Ushiku T, Motoi T et al. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am J Surg Pathol 2012;36:423–431.
Eversole LR, Leider AS, Nelson K . Ossifying fibroma: a clinicopathologic study of sixty-four cases. Oral Surg Oral Med Oral Pathol 1985;60:505–511.
Gollin SM, Storto PD, Malone PS et al. Cytogenetic abnormalities in an ossifying fibroma from a patient with bilateral retinoblastoma. Genes Chromosomes Cancer 1992;4:146–152.
Dal Cin P, Sciot R, Fossion E et al. Chromosome abnormalities in cementifying fibroma. Cancer Genet Cytogenet 1993;71:170–172.
Sawyer JR, Tryka AF, Bell JM et al. Nonrandom chromosome breakpoints at Xq26 and 2q33 characterize cemento-ossifying fibromas of the orbit. Cancer 1995;76:1853–1859.
de Mesquita Netto AC, Gomez RS, Diniz MG et al. Assessing the contribution of HRPT2 to the pathogenesis of jaw fibrous dysplasia, ossifying fibroma, and osteosarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol 2013;115:359–367.
Zhang TH, Liu HC, Liao GQ et al. Detection of Notch signaling molecules in cemento-ossifying fibroma of the jaws. J Oral Pathol Med 2010;39:263–268.
El-Mofty S . Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinicopathologic entities. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002;93:296–304.
Makek M . Clinical pathology and differential diagnosis of fibro-osseous lesions of the maxillofacial area—new aspects. Dtsch Z Mund Kiefer Gesichtschir 1986;10:212–217.
Makek MS . So called ‘fibro-osseous lesions’ of tumorous origin. Biology confronts terminology. J Craniomaxillofac Surg 1987;15:154–167.
Manjunatha BS, Das N, Naik S R G.. Trabecular variant of juvenile aggressive ossifying fibroma of anterior mandible. Pediatr Rep 2012;4:24.
Triantafillidou K, Venetis G, Karakinaris G et al. Ossifying fibroma of the jaws: a clinical study of 14 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2012;114:193–199.
MacDonald-Jankowski DS . Fibro-osseous lesions of the face and jaws. Clin Radiol 2004;59:11–25.
Abramowicz S, Goldwaser BR, Troulis MJ et al. Primary jaw tumors in children. J Oral Maxillofac Surg 2013;71:47–52.
Weaver J, Downs-Kelly E, Goldblum JR et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol 2008;21:943–949.
Ohta M, Seto M, Ijichi H et al. Decreased expression of the RAS-GTPase activating protein RASAL1 is associated with colorectal tumor progression. Gastroenterology 2009;136:206–216.
McLaughlin SK, Olsen SN, Dake B et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell 2013;24:365–378.
Robinson RA, Vincent SD . Tumors and Cysts of the Jaws. AFIP Series 4 Vol. 16. American Registry of Pathology, 2012, p 288.
Fletcher CD, Bridge JA, Hogendoorn P et al. World Health Organization Classification of Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2013, p 468.
Parham DM, Bridge JA, Lukacs JL et al. Cytogenetic distinction among benign fibro-osseous lesions of bone in children and adolescents: value of karyotypic findings in differential diagnosis. Pediatr Dev Pathol 2004;7:148–158.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Tabareau-Delalande, F., Collin, C., Gomez-Brouchet, A. et al. Chromosome 12 long arm rearrangement covering MDM2 and RASAL1 is associated with aggressive craniofacial juvenile ossifying fibroma and extracranial psammomatoid fibro-osseous lesions. Mod Pathol 28, 48–56 (2015). https://doi.org/10.1038/modpathol.2014.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.80
This article is cited by
-
Genomic Profiling of the Craniofacial Ossifying Fibroma by Next-Generation Sequencing
Head and Neck Pathology (2023)
-
Die neue WHO-Klassifikation der Kiefertumoren
Die Pathologie (2023)
-
MDM2 Gene Amplification and Expression of MDM2 and CDK4 are Rare in Ossifying Fibroma of Craniofacial Bones
Head and Neck Pathology (2022)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Odontogenic and Maxillofacial Bone Tumours
Head and Neck Pathology (2022)
-
Ossifying Fibromas of the Head and Neck Region: A Clinicopathological Study of 45 Cases
Head and Neck Pathology (2022)
