
Abstract
The molecular determinants involved in the progression of myxoid liposarcoma to increased cellularity/round cell change are poorly understood. We studied the PI3K/Akt pathway in myxoid and round cell liposarcomas using a tissue microarray composed of 165 tumors from 111 patients, and mutational analysis of PIK3CA in 44 cases. Activating PIK3CA mutations were found in 6/44 cases, 14%; mutations were more frequent in round cell vs myxoid tumors (5/15, 33% vs 1/29, 3%; P=0.013). Complete loss of PTEN, an alternative mechanism for PI3K/Akt activation, was found in 13/111 (12%) cases and was mutually exclusive with PIK3CA mutation. Strong IGF1R expression was demonstrated in 14/39 (36%) of round cell and 11/58 (19%) of myxoid tumors (P=0.062). Activation of the PI3K pathway was confirmed using immunohistochemical analysis for downstream targets phospho-S6 ribosomal protein and phospho-4EBP1. Phospho-4EBP1 was increased in round cell tumors compared with myxoid tumors (24/30, 80% vs 25/44, 57%; P=0.038) or tumors with treatment effect (10/24, 42%; P=0.02). Phospho-S6 was highly expressed in both myxoid and round cell tumors (29/47, 62% and 14/30, 47%, respectively; P=0.2). In tumors with PIK3CA mutation, any IGF1R expression, or loss of PTEN expression, phospho-4EBP1 was more frequently elevated compared with tumors without a known activating event in the PI3K pathway (55/72; 76% vs 3/8, 38%; P=0.033). These findings suggest that activation of the PI3K/Akt pathway via activating mutation of PIK3CA, loss of PTEN, or IGF1R expression have a role in round cell transformation. The PI3K/Akt pathway may therefore provide a therapeutic target in round cell liposarcoma.
Similar content being viewed by others

Main
Myxoid liposarcoma is one of the more common soft tissue sarcomas, comprising ∼33% of all liposarcomas and ∼10% of all adult soft tissue sarcomas.1 Tumors fall along a morphologic spectrum from paucicellular purely myxoid tumors to the more aggressive highly cellular round cell liposarcomas.2, 3, 4, 5 Both myxoid and round cell liposarcoma are characterized by chromosomal translocation, most frequently t(12;16) (FUS-DDIT3) or less commonly t(12;22) (EWSR1-DDIT3).2, 6 The fusion oncoproteins thus created are thought to have a role in tumor initiation.7, 8, 9 However, molecular factors associated with tumor progression, in particular round cell transformation, are poorly understood.
Recently, evidence has indicated a role for the PI3K/Akt pathway in myxoid to round cell progression. In carcinomas, the PI3K/Akt pathway is well characterized (Figure 1), and is known to be positively regulated by increased expression of the tyrosine kinase growth factor receptor IGF1R,10 activating mutations in PIK3CA,11, 12 as well as by loss of PTEN, a lipid and protein phosphatase serving as an inhibitory factor.13 Signaling through tyrosine kinase growth factor receptors via PI3K leads to phosphorylation of downstream intermediaries, including Akt and mTOR, and subsequent phosphorylation of targets such as S6 ribosomal protein and 4EBP1.14

Simplified PI3K/Akt pathway. Binding of growth factor to a receptor tyrosine kinase, such as IGF1R, activates PI3K, which phosphorylates a signaling molecule, phospatidyl inositol (PIP2), to its active form (PIP3). This in turn triggers a subsequent signaling cascade via Akt that results in phosphorylation of downstream targets such as S6 and 4EBP1. PI3K signaling is inhibited by PTEN, which dephosphorylates PIP3 to PIP2, thereby returning it to the inactive state.
In myxoid liposarcoma, overexpression of IGF1R has been reported to be associated with aggressive behavior and poor prognosis.15 While Akt is constitutively expressed in both myxoid and round cell liposarcoma, it was more frequently found in its activated (phosphorylated) form in round cell tumors.16 Moreover, PIK3CA mutations have recently been reported in 18% of myxoid and round cell liposarcoma; these mutations were associated with poor outcome.17 PIK3CA mutations most frequently involved hotspots in the helical and kinase domains, specifically in exons 9 and 20. These same mutations are also seen in a variety of carcinomas,12 where they are associated with activation of the PI3K/Akt pathway.18, 19
We hypothesized that activation of the PI3K/Akt pathway could be an important factor in progression of myxoid to round cell liposarcoma. We therefore used immunohistochemistry on a myxoid liposarcoma tissue microarray and PIK3CA mutational analysis to assess functional activation of the PI3K/Akt pathway, as well as the possible mechanism of this activation.
Materials and methods
Patients and Tumor Tissues
Acquisition of tissue specimens and clinical information were approved by the institutional review board of The University of Texas MD Anderson Cancer Center (UTMDACC), and performed in accordance with the Health Insurance Portability and Accountability Act regulations.
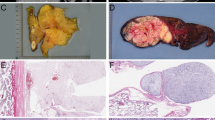
Formalin-fixed, paraffin-embedded myxoid and round cell liposarcoma specimens with available tissue blocks collected between 1987 and 2009 were retrieved from UTMDACC pathology archives, amounting to 165 unique tumors from 111 patients. All specimens were screened by at least two experienced soft tissue pathologists (Wang and/or Lazar as well as the primary clinical diagnosis). Only cases with classical histologic features of myxoid liposarcoma, consisting of small ovoid to round cells embedded in a prominent myxoid stroma characterized by a delicate plexiform vascular network, or round cell liposarcoma, with back-to-back primitive round cells with high nuclear: cytoplasmic ratios comprising >5% of tissue volume were included.1 Treated cases with no areas of classic histology were included only if diagnosis was confirmed by prior biopsy or recurrent disease. For the purpose of this study, tumors were classified as (1) purely myxoid, (2) containing areas of increased cellularity (round cell) change, or (3) showing therapeutic effect in the form of either adipocytic maturation, or extensive hyalinization with scattered residual tumor cells. Only those with confirmed myxoid or round cell histology and containing tissue adequate for analytic purposes were included. These specimens comprised 43 biopsies and 127 resection specimens. An additional 18 tumors with unusual morphology (marked spindling, pleomorphism),20 or where the diagnosis could not be definitively made on histologic review were excluded from the study, as were 10 cases with no viable tumor. Clinical information including demographic, tumor, and therapeutic information was retrieved from patient medical records and from the UTMDACC soft tissue tumor database, and tabulated for correlative analysis. Sites of primary and metastatic tumors were categorized as extremity (leg or arm), superficial trunk (chest wall, back, buttock, flank), retroperitoneum/intraabdominal, lung, bone, or paraspinal.
Tissue Microarray Construction
Hematoxylin and eosin (H&E)-stained sections were reviewed to define areas of pure myxoid or round cell change. An automated tissue microarray apparatus (ATA-27, Beecher Instruments, Sun Prairie, WI, USA) was used, and 0.2 cm punch samples (two per case, 330 total) were obtained from the tumor blocks and formatted into four recipient blocks. In addition, six cases (two paired punches/case) of normal fat, three atypical lipomatous tumors, two myxomas, and two pleomorphic liposarcomas were included for comparison. H&E staining of 4 μm tissue microarray sections was used to verify all samples.
Immunohistochemical Analysis
Immunohistochemical staining was performed on tissue microarray sections, using 4-μm-thick sections. Antigen retrieval was performed with 10 mM sodium citrate buffer, pH 6.0, for 10 min at 98 °C. An automated stainer (Ventana Medical Systems, Tucson, AZ, USA) was used following the manufacturer's instructions, and commercially available antibodies against PTEN (clone 6H2.1, Dako, Carpenteria, CA, USA; dilution 1:100), IGF1R (clone G11, Ventana; dilution 1:2 (prediluted)), phosphorylated 4EBP1 (p4EBP1) (Thr70, 9455, Cell Signaling, Danvers, MA, USA; dilution 1:100–1:200), phosphorylated S6 ribosomal protein (pS6) (Ser235/Ser236, 2211, Cell Signaling; dilution 1:100–1:200). Positive and negative controls were run in parallel. Sections were counterstained with hematoxylin. Labeling intensity for phosphorylated proteins was graded as high when tumor cells demonstrated diffuse, intense positive staining, and low when only weak staining was present. PTEN was considered to be negative when there was no staining in any tumor cells or weakly present only in rare tumor cells (constituting <10% of tumor mass), and positive if there was expression in ≥10% of cells (the staining was most often uniformly positive or negative). Tumor-associated vessel endothelium was used as an internal positive control. IGF1R staining was graded as negative (staining in <10% of tumor cells)=0, weak, predominately cytoplasmic=1, or strong, predominately membranous=2. Both p4EBP1 and pS6 were graded as negative=0, weak=1, moderate=2, strong=3.
PIK3CA Genotyping
DNA was extracted from two 20-μm-thick formalin-fixed paraffin-embedded tissue rolls cut from blocks with at least 80% tumor, using the QIamp DNA mini kit DNA isolation kit (Qiagen, Valencia, CA, USA). Polymerase chain reaction (PCR) used primers PIK3CAex9F 5′-TGACAAAGAACAGCTCAAAGC-3′, PIK3CAex9R 5′-TTAGCACTTACCTGTGACTCCA-3′, PIK3CAex20F 5′-TGATGACATTGCATACATTCG-3′ and PIK3CA20R 5′-TGTGTGGAAGATCCAATCCA-3′ with appropriate controls. PCR products (exon 9, 98 base pairs; exon 20, 136 base pairs) were detected by gel electrophoresis in 2% agarose and amplicon bands purified using QIAquick gel extraction kit (Qiagen). Direct sequencing used the above primers for exon 9 or 20, respectively, ABI Prism dye terminator sequence ready reaction kit, and ABI prism 3100-Avant genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Cell Culture and Mutational Analysis
Myxoid liposarcoma cell lines 402-91 and 1765-92 and primary cell cultures L1187, L1357, L1434, and L2187, derived from two round cell and two myxoid liposarcomas, respectively were cultured as previously described.21 Mutation analysis was performed on DNA extracted from cell lines as well as from corresponding frozen tumor tissue for primary cultures. Mutation analysis was performed for H1047R, E545K, and E542K in PIK3CA using TaqMan assay as previously described.22 PTEN immunohistochemistry was performed using whole sections of the corresponding paraffin blocks of the primary cultures.
Statistical Methods
Associations between histopathological features and immunophenotype were examined using the χ2 test or Fisher's exact test where appropriate.
Results
Demographics
The study population consisted of 111 patients, including 68 men and 43 women (Table 1). Age at presentation ranged from 17 to 79 years (median 42). Primary tumors mainly occurred in the lower extremity (84%), with trunk (12%) and upper extremity (4%) comprising the remainder. Metastatic disease most frequently occurred in the retroperitoneum (34%), while other sites of metastasis included chest wall (20%), lung (16%), bone (14%), paraspinal area (13%), and flank (5%). In all, 74 tumors were resected after treatment with either chemotherapy (n=35), radiation therapy (n=13), or both chemotherapy and radiotherapy (n=26).
Histopathological Characteristics
In all, 165 tumors were included in the study, comprising 82 primary tumors, 56 metastases, and 27 recurrent tumors (Table 2). There were 75 purely myxoid tumors, 43 tumors with areas of increased cellularity (round cell liposarcoma), and 45 tumors with histologic evidence of response to prior therapy in the form of adipocytic maturation or extensive hyalinization. Tumors represented on core biopsy tissue was more frequently from primary (29/43, 67% vs 54/127, 43%, P=0.0047) and untreated tumors (40/43, 93% vs 53/127, 42%, P<0.0001) compared with resection specimens. Complete or partial immunohistochemical profiles following tissue microarrray analysis were compiled for 58 myxoid tumors, 36 round cell tumors, and 37 tumors with therapy effect. Loss of tissue on the tissue microarray, uninterpretable staining artifact, or poor fixation, resulting in failure of internal controls to stain was most frequently seen in tissue core samples obtained from core needle biopsies and in older tissue specimens. These cases (n=34) were excluded from subsequent analyses.
PIK3CA Mutation Status
We first sought to confirm the prevalence of the most commonly reported point mutations in PIK3CA. Adequate tissue was available in 45 cases, including 18 myxoid, 10 round cell, and 17 tumors with treatment effect. Mutation status of two myxoid liposarcoma cell lines (402-91 and 1765-92), and four primary cell cultures was also investigated. Sufficient DNA was extracted in all but one case of myxoid liposarcoma. Mutations were found in 6/44, 14% of cases (Figure 2), and most frequently involved exon 20 (H1047R, four cases; G1049R, one case) (Table 3). One mutation in exon 9, E542K was identified. Four mutations involved round cell tumors and two involved treated cases. No mutations were found in myxoid tumors (P=0.012). We then reclassified treated tumors by their reported pretreatment histology as myxoid (n=12) or round cell (n=5). By this analysis, PIK3CA mutations were present in 1/29 (3%) of total myxoid tumors and 5/15 (33%) of round cell tumors (P=0.013). No mutation was identified in either myxoid liposarcoma cell line, or any of the four primary cell cultures.

PIK3CA exons 9 and 20 sequencing was performed in 44 patients on the TMA. Examples of Sanger sequencing for exons 9 and 20 mutations are displayed. (a) Exon 9, E542K. (b) Exon 20, H1047R. H1047R was the most frequently seen PIK3CA mutation in our cohort. The mutated base pair is denoted by black arrow on DNA sequence tracing, and codon change under affected codon (underlined in black). Reverse sequence pictured for (a), and forward sequence for (b). The non-mutated wild-type allele serves as an internal control.
Loss of PTEN
An alternative mechanism of activation of the PI3K/Akt pathway is loss of PTEN. Immunohistochemical studies were used to assess for the presence or absence of PTEN protein (Figure 3). There was complete loss of PTEN in 13/111, 12% of all cases. While no loss of PTEN was seen in tumors exhibiting treatment effect (0%, P=0.038 vs tumors without treatment effect), complete loss of PTEN was seen in 6/33, 18% of round cell tumors and 7/49 (14%) of myxoid tumors. Also, one of the primary cultures (L1434) demonstrated loss of PTEN protein expression.

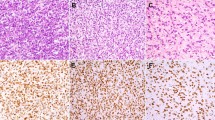
Immunohistochemical assessment of PTEN expression in myxoid and round cell liposarcoma. (a) Round cell liposarcoma with retained expression. (b) Myxoid liposarcoma with loss of PTEN. Vascular endothelium and scattered mast cells show retained PTEN expression, while tumor cells are negative. (Left, × 40, right × 200, original magnification).
IGF1R Expression
To determine if IGF1R could be involved in round cell transformation, we assessed IGF1R protein expression using immunohistochemistry (Figure 4). Strong staining was more frequent in round cell tumors compared with myxoid tumors and trended toward significance (14/39, 38% vs 11/58, 19%; P=0.062). However, weak staining was present in both round cell and myxoid tumors (13/39, 33% vs 25/58, 43%; P=0.33). In contrast, tumors with treatment effect were less likely to have either strong (1/34, 3%; P=0.004) or weak staining (3/34, 9%; P=0.001) compared with tumors without treatment effect.

IGF1R in myxoid and round cell liposarcoma. (a) Negative IGF1R immunohistochemical stain in myxoid liposarcoma. (b) Weak and (c) Strong IGF1R expression in round cell liposarcomas. (Left, × 40, right × 200, original magnification).
Phosphorylation of Downstream Targets of PI3K Pathway
In order to confirm activation of the PI3K/Akt pathway, we next assessed levels of two phosphorylated downstream targets of the PI3K pathway, 4EBP1 and S6 ribosomal protein (Figure 5). PS6 ribosomal protein and p4EBP1 were present in 67/107 (63%) and 59/99 (60%) of all cases, respectively. High expression of pS6 was seen in 29/47 (62%) of myxoid tumors, 14/30 (47%) of round cell tumors, and 24/30 (80%) of tumors with evidence of treatment effect. There was no significant difference in pS6 expression between myxoid and round cell tumors.

Phospho-S6 and 4EBP1 expression in myxoid and round cell liposarcoma. (a, b) Immunohistochemical stains demonstrating low and high expression of pS6, respectively. (c, d) Low and high expression of p4EBP1, respectively. ( × 40, original magnification).
In contrast, high levels of p4EBP1 were more frequently found in round cell liposarcoma compared with myxoid liposarcoma (24/30, 80% vs 25/44, 57%; P=0.038), while tumors with treatment effect were least likely to demonstrate high levels of p4EBP1 (10/24, 42%; P=0.021). Of note, there were no significant differences in either pS6 or p4EBP1 expression between treated myxoid or round cell tumors in areas without marked histologic evidence of response when compared with untreated tumors (data not shown).
Correlation of IGF1R Expression, PIK3CA Mutation and PTEN Status with pS6 and p4EBP1
There were 31 cases with complete data on PIK3CA mutational status, PTEN loss, and IGF1R expression. No cases with PIK3CA mutation also had loss of PTEN. In contrast, weak cytoplasmic IGF1R expression was seen in two cases with loss of PTEN and one case with PIK3CA mutation, and strong IGF1R expression in one case with PIK3CA mutation.
There were 72 cases with any activating event in the PI3K/Akt pathway (PTEN loss, PIK3CA mutation, IFGR1 expression) and nine cases with no activating event (and complete data on all three activating factors). In cases with an activating event, 55/72, 76% had high p4EBP1, vs 3/8, 38% of cases with no activating event (P=0.033), while 43/72, 60% had high pS6 vs 6/9, 67% of cases with no activating event (P=0.74).
In order to adjust for confounding effects of treatment response, we then excluded tumors with treatment effect and compared tumors with myxoid or round cell histology only and no hyalinization or adipocytic maturation. Unfortunately, complete data on all three activating events were not available in many cases; therefore, for the purposes of this analysis, tumors with known negativity in 2/3 factors, with the third (usually PIK3CA mutational status) unknown, were considered to be negative for an activating event. In these cases, presence of an activating event was significantly associated with high p4EBP1 (51/67, 76%, vs 5/14, 36% P=0.005), but not with high pS6 (39/67, 58% vs 5/14, 36% P=0.12). Thus, even in the absence of treatment effect, high pS6 was not significantly associated with activation of the PI3K pathway.
Discussion
We demonstrated that while evidence of PI3K pathway activation is seen in both myxoid and round cell liposarcoma, such activation is significantly associated with round cell change. Tumors with round cell change were more likely to have strong membranous IGF1R expression or an activating mutation in PIK3CA than purely myxoid tumors. Moreover, activation of the PI3K/Akt pathway in round cell tumors was verified by the increased expression of p4EBP1 compared with myxoid tumors. Increased p4EBP1 in both myxoid and round cell tumors was significantly associated with activating events upstream in the PI3K/Akt pathway (IGF1R expression, PTEN loss, or PIK3CA mutation). Moreover, PIK3CA mutation and PTEN loss are mutually exclusive events and were usually not seen in conjunction with increased IGF1R expression. Taken together, these findings support a role for increased flux through the PI3K/Akt pathway in round cell change, a form of tumor progression in myxoid liposarcoma. Our findings confirm and expand upon the findings of a previous high-throughput study demonstrating PIK3CA mutations and activation of the PI3K/Akt pathway in myxoid and round cell liposarcoma.17
In addition, we found differences in IGF1R, PTEN, p4EBP1, and pS6 expression between tumors with treatment response and tumors with myxoid or round cell morphology. One limitation of this finding is the paucity of viable tumor cells in treated tumors with extensive hyalinization. Interpretation may have been skewed by the presence of non-viable tumor cells or infiltrating cells such as histiocytes mimicking tumor cells. Nevertheless, the difference in p4EBP1 expression remained significant even when comparing only treated tumors with adipocytic maturation vs myxoid/round cell tumors. This suggests that the PI3K/Akt pathway is down-regulated in maturing tumor cells in response to therapy. However, residual foci of myxoid or round cell histology within treated tumors showed no significant difference in expression of pS6 or p4EBP1 compared with untreated tumors. The driving forces behind the response to therapy or lack thereof have yet to be elucidated.
Previously, Barretina et al17 described the clustering of PIK3CA mutations in myxoid liposarcoma in exons 9 and 20, most frequently involving codons 542, 545, and 1047, with mutations appearing in 13/71 (18%) of tumors. We found a similar percentage of mutations in our cohort (6/44 (13%)), although our analysis was limited to the two hotspots in exons 9 and 20 and could not identify the remainder of the reported rare mutations elsewhere in the gene. Our cohort possessed fewer mutations in the helical domain (exon 9) and more in the kinase domain (exon 20) than were previously described. Of note, in the prior study of myxoid liposarcoma, H1047R mutations were reportedly not associated with increased pAkt; this was postulated to be due to high-level PTEN expression in these tumors.17 We were unable to directly assess Akt phosphorylation status, as it was not robustly detectable in the formalin-fixed paraffin-embedded tissues used in this study. Nor did we find higher PTEN levels in tumors with H1047R mutation. This discrepancy may be due to the method of analysis (tissue immunohistochemistry vs western blot). Nevertheless, the functionality of PIK3CA mutations in our cohort is supported by elevated levels of p4EBP1 in these cases.
PIK3CA mutation was previously reported to be associated with shortened disease-specific survival. While the authors did not report on myxoid vs round cell histology,17 their finding would be in keeping with our discovery that PIK3CA mutations are more frequent in the more aggressive round cell tumors. Increased pAkt has also been reported in round cell tumors,16 lending additional support to a role for the PI3K pathway in driving round cell transformation and tumor aggression.
We assessed two downstream targets of the PI3K pathway in order to confirm pathway activation. 4EBP1 is a translational repressor, known to be a direct target of the mTOR complex, and is inactivated by phosphorylation,14, 23 while phosphorylation of S6 ribosomal protein is thought to regulate protein translation.24, 25 Previous reports have indicated that S6 and 4EBP1 are equivalent markers of PI3K activation.26, 27, 28 However, we found that only p4EBP1 was associated with round cell liposarcoma or activating event in the PI3K/Akt pathway, while pS6 was only slightly elevated in these tumors. In fact, pS6 was most frequently seen at high levels in tumors with treatment effect, which were least likely to have evidence of an activating event in the PI3K pathway. One possible explanation for this finding is that is that S6 may be phosphorylated in tumors with treatment effect as part of the anti-apoptotic role of the PI3K pathway—a role that is known to involve the p70 S6 kinase responsible for the majority of S6 phosphorylation.29, 30 Thus, elevated pS6 in treated tumors could represent cellular response to stress with activation of survival factors rather than an indicator of proliferation and transformation. Consistent with this theory, high levels of pS6 and p4EBP2 were each seen in 36% of tumors in our cohort without evidence of treatment effect and no known activating event in the PI3K pathway. Increased levels of both pS6 and p4EBP1 were then seen in tumors with activating events, although the change in expression only reached significance with p4EBP1. These findings suggest that in tumors without treatment effect, phosphorylation of S6 and 4EBP1 are both mediated by the PI3K pathway. However, the presence of phosphorylated S6 and 4EBP1 in 36% of tumors without IGF1R/PIK3CA mutation/PTEN loss suggests that additional mechanisms of increased flux through the PI3K/Akt pathway are present in myxoid and round cell liposarcoma. Other activated receptor tyrosine kinases, including PDGFRB and EGFR, have been identified in myxoid liposarcoma,16 which may also lead to increased PI3K signaling. Additional studies are needed to further elucidate the myriad mechanisms by which PI3K/Akt signaling may be activated in these tumors.
No PIK3CA mutations were identified in two cell lines or four primary cell cultures from myxoid and round cell liposarcomas, although one of four primary cultures was found to have loss of PTEN expression. These cell lines and primary cultures could be used to model the role of the PI3K/Akt pathway in round cell transformation in myxoid liposarcoma, and to better understand the potential role of this pathway in tumor proliferation and invasion. Moreover, the primary cell culture with PTEN loss, and therefore activation of the PI3K pathway, may represent a valuable model in which to assess the efficacy of PI3K/Akt pathway inhibitors such as rapamycin or temsirolimus in treating these tumors. Additional study is needed to determine if the other cell lines and primary cultures may have other activating events in the PI3K pathway.
In summary, our findings posit a role for activation of the PI3K/Akt pathway in round cell transformation of myxoid liposarcoma. Mechanisms of activation include increased IGF1R, activating mutations in PIK3CA, as well as loss of PTEN. Preferentially increased levels of p4EBP1 are seen in round cell tumors, while pS6 may be affected by additional factors. Additional studies are needed to further elucidate the role of PI3K pathway activation in driving round cell transformation. If our data are confirmed, they suggest that targeted therapy against factors involved in the PI3K/Akt pathway, such as mTOR, could someday have a role in the treatment of aggressive round cell liposarcoma.
References
Antonescu C, Ladanyi M . Myxoid liposarcoma. In: Fletcher CDM, Unni KK, Mertens F (eds). Pathology and Genetics of Tumors of Soft Tissue and Bone. IARC Press: Lyon, 2002, pp 40–43.
Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res 2001;7:3977–3987.
Evans HL . Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol 1979;3:507–523.
Smith TA, Easley KA, Goldblum JR . Myxoid/round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol 1996;20:171–180.
Kilpatrick SE, Doyon J, Choong PF, et al. The clinicopathologic spectrum of myxoid and round cell liposarcoma. A study of 95 cases. Cancer 1996;77:1450–1458.
ten Heuvel SE, Hoekstra HJ, van Ginkel RJ, et al. Clinicopathologic prognostic factors in myxoid liposarcoma: a retrospective study of 49 patients with long-term follow-up. Ann Surg Oncol 2007;14:222–229.
Riggi N, Cironi L, Provero P, et al. Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res 2006;66:7016–7023.
Kuroda M, Ishida T, Takanashi M, et al. Oncogenic transformation and inhibition of adipocytic conversion of preadipocytes by TLS/FUS-CHOP type II chimeric protein. Am J Pathol 1997;151:735–744.
Perez-Losada J, Sanchez-Martin M, Rodriguez-Garcia MA, et al. Liposarcoma initiated by FUS/TLS-CHOP: the FUS/TLS domain plays a critical role in the pathogenesis of liposarcoma. Oncogene 2000;19:6015–6022.
Tao Y, Pinzi V, Bourhis J, et al. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway—therapeutic perspectives in cancer. Nat Clin Pract Oncol 2007;4:591–602.
Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 2004;3:772–775.
Samuels Y, Velculescu VE . Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004;3:1221–1224.
Vivanco I, Sawyers CL . The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489–501.
Carrera AC . TOR signaling in mammals. J Cell Sci 2004;117:4615–4616.
Cheng H, Dodge J, Mehl E, et al. Validation of immature adipogenic status and identification of prognostic biomarkers in myxoid liposarcoma using tissue microarrays. Hum Pathol 2009;40:1244–1251.
Negri T, Virdis E, Brich S, et al. Functional mapping of receptor tyrosine kinases in myxoid liposarcoma. Clin Cancer Res 2010;16:3581–3593.
Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet 2010;42:715–721.
Yuan TL, Cantley LC . PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008;27:5497–5510.
Miled N, Yan Y, Hon WC, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007;317:239–242.
Alaggio R, Coffin CM, Weiss SW, et al. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 2009;33:645–658.
Willems SM, Schrage YM, Bruijn IH, et al. Kinome profiling of myxoid liposarcoma reveals NF-kappaB-pathway kinase activity and casein kinase II inhibition as a potential treatment option. Mol Cancer 2010;9:257.
van Eijk R, Licht J, Schrumpf M, et al. Rapid KRAS, EGFR, BRAF and PIK3CA mutation analysis of fine needle aspirates from non-small-cell lung cancer using allele-specific qPCR. PLoS One 2011;6:e17791.
Gingras AC, Kennedy SG, O'Leary MA, et al. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 1998;12:502–513.
Stewart MJ, Thomas G . Mitogenesis and protein synthesis: a role for ribosomal protein S6 phosphorylation? Bioessays 1994;16:809–815.
Meyuhas O . Physiological roles of ribosomal protein S6: one of its kind. Int Rev Cell Mol Biol 2008;268:1–37.
Iwenofu OH, Lackman RD, Staddon AP, et al. Phospho-S6 ribosomal protein: a potential new predictive sarcoma marker for targeted mTOR therapy. Mod Pathol 2008;21:231–237.
Cen L, Arnoczky KJ, Hsieh FC, et al. Phosphorylation profiles of protein kinases in alveolar and embryonal rhabdomyosarcoma. Mod Pathol 2007;20:936–946.
Dobashi Y, Suzuki S, Sato E, et al. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod Pathol 2009;22:1328–1340.
Harada H, Andersen JS, Mann M, et al. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 2001;98:9666–9670.
Downward J . PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol 2004;15:177–182.
Acknowledgements
This work was supported in part by National Cancer Institute at the National Institutes of Health (RO1CA138345 Grant to DL) and the MD Anderson Physician-Scientist Program (AJL). We greatly appreciate the philanthropic support of the Lobo, Margolis, and Jackson families toward our liposarcoma research program. We are grateful to Professor Dr Pierre Aman (Lundberg Laboratory for Cancer Research (LLCR), Department of Pathology, Sahlgrenska Academy at University of Gothenburg, Gothenburg, Sweden) for providing myxoid liposarcoma cell lines 402-91 and 1765-92.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Demicco, E., Torres, K., Ghadimi, M. et al. Involvement of the PI3K/Akt pathway in myxoid/round cell liposarcoma. Mod Pathol 25, 212–221 (2012). https://doi.org/10.1038/modpathol.2011.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.148
Keywords
This article is cited by
-
Myxoid Liposarcomas: Systemic Treatment Options
Current Treatment Options in Oncology (2023)
-
Myxoid pleomorphic liposarcoma is distinguished from other liposarcomas by widespread loss of heterozygosity and significantly worse overall survival: a genomic and clinicopathologic study
Modern Pathology (2022)
-
Molecular signatures of tumor progression in myxoid liposarcoma identified by N-glycan mass spectrometry imaging
Laboratory Investigation (2020)
-
Establishment and characterization of a new human myxoid liposarcoma cell line (DL-221) with the FUS-DDIT3 translocation
Laboratory Investigation (2016)
-
Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis
Oncogene (2016)
