
Abstract
The diagnostic criteria for acute erythroid leukemia have been controversial since this disease was initially described. Using the current World Health Organization classification criteria, we retrospectively reviewed cases of acute myeloid leukemia or myelodysplastic syndrome in which erythroid precursors were ≥50% of the bone marrow nucleated cell population and the diagnosis of erythroleukemia was considered using older classification schemes. We collected 90 cases and separated them into four diagnostic groups: acute erythroid leukemia, erythroleukemia or erythroid/myeloid type (n=20); acute myeloid leukemia with myelodysplasia-related changes (n=22); therapy-related acute myeloid leukemia (n=32); and refractory anemia with excess blasts and preceding or concurrent history of erythropoietin therapy for anemia (n=16). Patients with acute erythroid leukemia were the youngest patient group and had the best overall survival. There was a statistically significant difference in overall survival between patients with acute erythroid leukemia versus acute myeloid leukemia with myelodysplasia-related changes (P=0.003) and between patients with acute erythroid leukemia versus therapy-related acute myeloid leukemia (P<0.0001). The presence of complex cytogenetic abnormalities (>3) was the only statistically significant independent variable that adversely affected survival in the acute erythroid leukemia group. Monosomy 5/del(5q) and monosomy 7/del(7q) were overrepresented in the context of complex chromosomal abnormalities. Our data suggest that acute erythroid leukemia, as currently defined in the World Health Organization classification, has become a rare disease. A majority of the cases reported previously as erythroleukemia are now classified as other entities. In addition, our data suggest that the current definition of acute erythroid leukemia, erythroleukemia type remains heterogeneous. One subset of acute erythroid leukemia patients has relatively low blast counts and are diploid. The prognosis of this patient subset is relatively good. The other subset has cytogenetic abnormalities similar to those in myelodysplastic syndromes and a poor prognosis.
Similar content being viewed by others


Main
Since its inception almost a century ago, the definition and prognosis of patients with leukemia of erythroid lineage has been controversial. Copelli, in 1912, was the first to describe ‘erythromatosis’.1 In 1917, Di Guglielmo described, under the designation ‘erythroleukemia’, a patient with proliferation of abnormal immature erythroid cells, myeloblasts and megakaryocytes. In 1923, he defined ‘acute erythremia’—a pure erythroid proliferation analogous to acute leukemia. Di Guglielmo continually stressed the ‘purity’ of the disorder as ‘an autonomous pathologic entity’ of the erythropoietic tissue.2 In 1940, Moeschlin definitively established the term ‘erythroleukemia’.3
In 1958, Dameshek and Baldini suggested the term ‘Di Guglielmo syndrome’ to encompass all aspects of erythroleukemia and to honor their colleague. They suggested that erythroleukemia passed through several stages: (1) a predominantly erythroblastic proliferation in the bone marrow; (2) a mixed erythroblastic–myeloblastic process; and (3) a predominantly myeloblastic neoplasm. They also stressed that Di Guglielmo syndrome is a proliferative disease of the bone marrow, thus suggesting the modern concept of a stem cell disorder.2 In 1969, Dameshek further developed the concept of erythroleukemia as ‘a self-perpetuating, myeloproliferative disorder of undetermined origin characterized by progressive anemia; striking erythroblastic hyperplasia of megaloblastic, megaloblastoid or normoblastic types; and the gradual development of increasing number of myeloblasts’ with frequent eventual progression to erythroleukemia and subsequently acute myeloid leukemia.4 Others also used the term ‘Di Guglielmo disease’ specifically for the disease first described by Di Guglielmo as ‘acute erythremia’.5
Concomitant with development of the concept of erythroleukemia, it became known that erythroleukemia could be accompanied by multilineage dysplasia.6, 7 Erythroleukemia was also shown to be characterized by various cytogenetic abnormalities without any consistent pattern, including aneuploidy (monosomies and trisomies) and complex chromosomal karyotypes.8, 9 In 1969, Heath et al10 confirmed the tendency of erythroleukemia toward hypodiploidy and, on the basis of similar cytogenetic abnormalities, suggested that erythroleukemia is a variant of acute myeloid leukemia.
The French–American–British Cooperative Group initially sought to avoid confusion between erythroleukemia, which they designated as M6, and other megaloblastic and dyserythropoietic states, by setting the overall percentage of myeloblasts and promyelocytes at 30% of all nucleated bone marrow cells.11 In 1985, the criteria for erythroleukemia were revised to require that at least 30% of nonerythroid cells be type I and type II blasts, and cases in which blasts made up <30% of nonerythroid cells were diagnosed as a myelodysplastic syndrome.12 The rationale behind the changing the calculation of the myeloblast percentage from that of total nonerythroid bone marrow cells was largely arithmetical: in cases in which erythroid precursors accounted for >70% of the bone marrow cellularity, it was impossible to establish the diagnosis of acute leukemia because blasts represented <30% of total bone marrow cells.
The 2001 World Health Organization classification of myeloid neoplasms incorporated all known aspects of these diseases including morphology, immunophenotype, genetic data and clinical features.13 In this classification, acute erythroid/myeloid leukemia or acute erythroleukemia was defined as acute myeloid leukemia in which erythroid precursors represented at least 50% of the nucleated bone marrow cell population with myeloblasts accounting for at least 20% of the nonerythroid cell population. The investigators suggested that cases meeting the criteria for both acute erythroid leukemia and acute myeloid leukemia with multilineage dysplasia should be diagnosed as ‘acute myeloid leukemia with multilineage dysplasia, acute erythroid/myeloid type’.13 In the 2008 update of the World Health Organization classification,14 neoplasms in which ≥50% of the bone marrow cells are erythroid precursors were further clarified, but some aspects of this classification remain unclear. Specifically, cases in which blasts account for fewer than 20% of leukocytes in the peripheral blood and <20% of the nonerythroid cells in the bone marrow are considered myelodysplastic syndrome. However, there remains no consensus as to whether a myelodysplastic syndrome should then be classified on the basis of blast percentage of all nucleated cells, or only nonerythroid bone marrow cells (ie, lymphocytes, plasma cells and megakaryocytes excluded). The current World Health Organization classification also makes acute erythroid leukemia, in part, a diagnosis of exclusion, as cases of acute myeloid leukemia with myelodysplasia-related changes, therapy-related acute myeloid leukemia or acute myeloid leukemia with maturation and increased erythroid precursors, and reactive erythroid hyperplasia after therapy or administration of erythropoietin must be excluded.
In this study, we applied the 2008 World Health Organization classification criteria to erythroid predominant cases of acute myeloid leukemia or myelodysplastic syndrome in our files. In many of these cases, the diagnosis of erythroleukemia or acute erythroid leukemia was suggested or established using older classification systems. Our results show that the current World Health Organization criteria substantially refine and reduce the frequency of acute erythroid leukemia. Nevertheless, cases of acute erythroid leukemia that remain classified as such are heterogeneous, with at least two subsets with different prognosis that correlate with the presence of cytogenetic abnormalities.
Materials and methods
Study Group
We searched the files of the Department of Hematopathology at MD Anderson Cancer Center from 1992 to April 2009. The criterion for inclusion into this study was a diagnosis of acute myeloid leukemia or myelodysplastic syndrome with erythroid predominance (≥50% of nucleated bone marrow cells) at time of initial presentation at our institution. The medical records of each patient were reviewed to obtain data such as age, sex, complete blood count, immunophenotype as determined by flow cytometry, cytogenetic results and molecular data. Cases were excluded if clinical or laboratory data were missing that precluded accurate diagnostic classification. Cases with a history of Janus kinase 2 mutation-positive myeloproliferative neoplasm with increased blasts in either accelerated or blast phase associated with erythroid predominance were also excluded. The Institutional Review Board of MD Anderson approved this study.
Morphologic Evaluation
Wright–Giemsa stained bone marrow aspirate smears and hematoxylin–eosin-stained bone marrow aspirate clot and trephine biopsy sections were reviewed. Bone marrow differential cell counts were performed on all cases. The presence or absence of dysplasia was determined and the percentage of dysplastic erythroid, granulocytic and megakaryocytic precursors in the bone marrow was evaluated as described by Goasguen et al.15 Bone marrow aspirate smears were evaluated by cytochemical analysis for myeloperoxidase and a-naphthyl butyrate esterase using methods described previously.16
The criteria of the 2008 World Health Organization classification were used to establish the diagnosis of acute myeloid leukemia.14 All cases in this study were erythroid rich (>50% of bone marrow nucleated cells). The study group was further subdivided into four subgroups as follows: acute erythroid leukemia; acute myeloid leukemia with myelodysplasia-related changes; therapy-related acute myeloid leukemia; and refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia. Patients with acute erythroid leukemia met the World Health Organization criteria for acute erythroid leukemia and had no history of myelodysplastic syndrome or therapy for another neoplasm. Patients with acute myeloid leukemia with myelodysplasia-related changes met the criteria for acute myeloid leukemia and either had a history of myelodysplastic syndrome or morphologic evidence of myelodysplasia, or had myelodysplastic syndrome-related cytogenetic abnormalities as specified in the World Health Organization classification.17 Patients with therapy-related acute myeloid leukemia had a history of another neoplasm treated with either radiation therapy, chemotherapy or both.18 Patients with refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia did not meet World Health Organization criteria for acute myeloid leukemia and had a known history or were on therapy with erythropoietin at time of bone marrow examination.
Flow Cytometry Immunophenotyping and Conventional Cytogenetics
Three- or four-color flow cytometry immunophenotypic analysis was performed on bone marrow aspirate specimens using a FACScan or FACSCalibur instrument, as has been described.16 Antibodies specific for the following antigens were used: CD2, CD3 (surface and cytoplasmic), CD5, CD7, CD10, CD13, CD14, CD15, CD19, CD20, CD33, CD34, CD38, CD41, CD45, CD56, CD64, CD117, HLA–DR, myeloperoxidase and terminal deoxynucleotidyl transferase. Blasts were gated for analysis using CD45 expression and light side scatter characteristics. Blasts were considered positive for a given antigen if positive cells constituted at least 20% of the blast population as compared with isotype control.
Conventional cytogenetic analysis was performed on bone marrow aspirate specimens using standard Giemsa trypsin G-banding procedures as described previously.19 The karyotypes at the time of initial diagnosis were classified as diploid, simple (≤3 cytogenetic abnormalities) or complex (>3 cytogenetic abnormalities).
Detection of Fms-Like Tyrosine Kinase 3, v-Kit Hardy–Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog, RAS Viral Oncogene Homolog and Janus Kinase 2 Mutations
Genomic DNA was extracted from fresh bone marrow aspirate specimens or archival paraffin-embedded tissue blocks. The fms-like tyrosine kinase 3 gene was assessed for internal tandem duplication and codon 835–836 point mutations in the activation loop of the tyrosine kinase domain by polymerase chain reaction assays followed by capillary electrophoresis.20 The forward primers were labeled with 6-carboxyfluorescein. For codon 835–836 point mutation analysis, polymerase chain reaction products were digested with EcoRV. In unmutated cases, digestion creates two fragments, of which the 80-bp fragment is labeled. In mutated cases, a 129-bp fragment is detected.
To detect Janus Kinase 2 mutations, exon 14-F (5′-GGACCAAAGCACATTGTATCCTC-3′) and exon 14-R (5′-GGGCATTGTAACCTTCTACTT-3′) were amplified by polymerase chain reaction according to standard protocols. The resulting 400-bp polymerase chain reaction product was purified by using the Qiagen polymerase chain reaction Purification Kit. Quantitative allele-specific suppressive polymerase chain reaction was performed on the purified 400-bp polymerase chain reaction product by using a sequence detection system 7000 platform (Applied Biosystems) as previously described.21
Sequence analysis of exons 8 and 17 of the v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog (C-KIT) gene was carried out on genomic DNA extracted from bone marrow aspirate samples according to previously reported.21
Polymerase chain reaction-based DNA pyrosequencing of v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-ras-2) and neuroblastoma RAS viral (v-ras) oncogene homolog (N-ras) was conducted in separate reactions for exon 1 (including codons 12 and 13) and exon 2 (including codon 61) as described previously.21
Statistical Analysis
Median age and peripheral blood and bone marrow indices were calculated. The Kaplan–Meier method was used to calculate survival fractions of each group. The log-rank (Mantel–Cox) test was used to compare paired survival curves between all the groups. Cases were stratified by age (≤65 or >65 years), evidence of multilineage dysplasia, degree of cytogenetic abnormality (diploid/simple versus complex), sex and evidence of relapse. The Bonferroni method was used to correct a P-value for multiple comparisons of overall survival among four subgroups. Specifically, a conventionally accepted P-value (0.05) was divided by K-value (equal to number of overall comparisons) to compute a Bonferroni corrected threshold. Only P-values that were less than Bonferroni corrected threshold were considered statistically significant.
Results
The study group included 90 cases with erythroid predominance classified originally as a myeloid neoplasm and thought to be consistent with or suggestive of erythroleukemia at our institution. Many of the older cases in this study were initially diagnosed as acute myeloid leukemia M6 type using the French–American–British classification12 or as acute erythroid/myeloid leukemia using the 2001 World Health Organization classification.13 Applying the 2008 World Health Organization classification criteria to this group, we recognized four subgroups: 20 cases of acute erythroid leukemia, erythroleukemia or erythroid/myeloid type; 22 cases of acute myeloid leukemia with myelodysplasia-related changes; 32 cases of therapy-related acute myeloid leukemia; and 16 cases of refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia. We also identified two cases of pure erythroid leukemia. These cases were excluded from this study because the slides were not available for one case and clinical data was incomplete for the other patient.
Patient age, sex, complete blood count, bone marrow differential count and survival are presented according to the four subgroups in Table 1 . In all subgroups, the major complete blood count finding at time of diagnosis was pancytopenia with absolute neutropenia. In all subgroups, the bone marrow was hypercellular for age with increased myeloblasts, erythroid predominance and marked depletion of granulocytic precursors. The conventional cytogenetic data for all subgroups are summarized in Table 2 . Blasts in the peripheral blood and bone marrow showed a myeloid immunophenotype by flow cytometric analysis. Aberrant CD7 expression was a common finding in acute myeloid leukemia: 28% of acute erythroid leukemia; 21% of acute myeloid leukemia with myelodysplasia-related changes; and 32% of therapy-related acute myeloid leukemia. Only 7% of refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia cases showed aberrant CD7 expression. The results of molecular genetic analyses are shown in Table 3 .
Acute Erythroid Leukemia
A diagnosis of acute erythroid leukemia, erytholeukemia (erythroid/myeloid) type was made in 20 patients. The median age of patients in this subgroup was 53 years (range, 12 to 76), which was younger than the other three subgroups. The male-to-female ratio was four to one. Anemia and thrombocytopenia were present in all patients. Pancytopenia with absolute neutropenia was present in 14 (70%) patients. Bone marrow was hypercellular for age in 14 (70%) cases. Increased myeloblasts, erythroid predominance and marked depletion of granulocytic precursors were observed in all cases. The blast count ranged from 6 to 37% (median, 16%) of all nucleated bone marrow elements, and was >20% of the nonerythroid elements in every case (range, 21–80%).
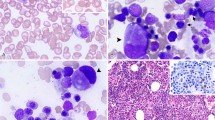
Dysplasia was present in all cases (Figure 1, Table 4 ). Dyserythropoiesis was present in 15 (75%) cases, with 1 (6%) case having >50% dysplastic erythroid elements. Increased ring sideroblasts (>15%) were observed in 7 of 14 (50%) cases with available iron stains. Dysgranulopoiesis was observed in 15 of 17 (88%) cases; 3 remaining cases had too few granulocytic precursors for adequate evaluation. Two cases had >50% dysplastic granulocytic precursors. Megakaryocytic dysplasia was present in 17 of 19 (89%) cases; in 1 case too few megakaryocytes were present for assessment. Eleven cases had >50% dysplastic megakaryocytes. Severe multilineage dysplasia (ie, dysplasia in at least 50% of the cells in at least two hematopoietic lineages was identified in seven cases, four of which showed a complex karyotype.

Dyserythropoiesis, dysgranulopoiesis and dysmegakaryopoiesis in acute erythroid leukemia. (a) Dysplastic normoblasts in bone marrow aspirate smear (Wright–Giemsa stain). (b) Bone marrow aspirate smear showing ring sideroblasts (Prussian blue followed by safranin O stain). (c) Bone marrow aspirate smear showing a dysplastic neutrophil (Wright–Giemsa stain). (d) Bone marrow biopsy specimen showing increased number of dysplastic megakaryocytes (hematoxylin–eosin stain).
The karyotype was diploid in 11 of the patients (Tables 2 and 4). Nine (45%) patients had an abnormal karyotype: non-complex abnormalities in 4 patients and complex in 5 patients. Unbalanced cytogenetic abnormalities were observed primarily in the setting of a complex karyotype. It is noteworthy that trisomy 8 was observed in the context of noncomplex karyotypes (≤3 abnormalities) in three of four cases. One patient who had a diploid karyotype and single (megakaryocytic) lineage dysplasia (67% megakaryocytes) had a distinctive family history. The father had acute leukemia at age 71 years; an uncle had acute leukemia at age 55 years; a cousin had acute leukemia at age 78 years; another cousin developed aplastic anemia at age 54 years; a second cousin had myelodysplastic syndrome at age 70 years; and a cousin had pancreatic carcinoma.
Acute Myeloid Leukemia with Myelodysplasia-Related Changes
There were 22 patients in this subgroup ranging from 36 to 80 years old, with a median age of 60 years. There was a 1.4:1 male predominance. Six patients had a history of myelodysplastic syndrome from 8 months to 7 years, before the diagnosis of acute myeloid leukemia with myelodysplasia-related changes. Dyserythropoiesis was observed in 21 (96%) cases, and was present in all 7 (27%) cases of trilineage dysplasia, all 11 (50%) cases of bilineage dysplasia, and in 3 (14%) cases as a single dysplastic lineage. Ringed sideroblasts were documented in one case. One case had no dysplasia in hematopoietic precursors and had a diploid karyotype, but was included in this subgroup based on a history of myelodysplastic syndrome and bone marrow with >20% blasts.
Cytogenetic results at the time of diagnosis were available for 21 patients (Table 5 ). The karyotype was complex in 12 of 21 (57%) cases; unbalanced cytogenetic abnormalities were observed almost exclusively in the context of a complex karyotype. Exceptions included one case of monosomy 7 associated with t(11;21)(q14;q12) and one case of del(20)(q12) associated with trisomy 8. Trisomy 8 was observed in the context of noncomplex karyotypes in two cases.
Therapy-Related Acute Myeloid Leukemia
There were 32 patients with therapy-related acute myeloid leukemia with a median age of 65 years (range, 21 to 78). The male-to-female ratio was two to one (Table 1). Nineteen patients had a history of a hematolymphoid neoplasm, whereas 13 patients had a history of a non-hematolymphoid neoplasm. All patients were treated with chemotherapy and/or a combination radiation and chemotherapy, with a mean interval of 7 years before the diagnosis of therapy-related acute myeloid leukemia. Dyserythropoiesis was observed in 30 of 31 (97%) cases evaluated, and was present in all 12 (39%) cases of trilineage dysplasia, in 11 (29%) cases of bilineage dysplasia, and as a single dysplastic lineage in 7 (13%) cases. Ringed sideroblasts were documented in three cases. One (3%) case had dysgranulopoiesis as the only manifestation of hematopoietic dysplasia.
Karyotypes were available for all 32 cases (Tables 2 and 6 ). The karyotype was complex in 19 of 32 (59%) cases. Unbalanced cytogenetic abnormalities were observed predominantly in the context of a complex karyotype. Trisomy 8 was observed in the context of complex cytogenetic abnormalities in three of four cases. A diploid karyotype was present in three (9%) cases.
Refractory Anemia with Excess Blasts with Preceding/Concurrent History of Erythropoietin Therapy
There were 16 patients in this subgroup, with erythroid predominance possibly attributable to erythropoietin administration. These patients constituted the oldest subgroup, with a median age of 67 years (range, 58–89). There was a seven to one male predominance (Table 1). All patients had a history of myelodysplastic syndrome, from 1 month to 3 years in duration, and were receiving regular injections of erythropoietin or its analog immediately before, or at the time of, bone marrow examination. Dyserythropoiesis was observed in all 16 (100%) cases, present in all 10 (63%) cases of trilineage dysplasia, in 4 (25%) cases of bilineage dysplasia, and as a single dysplastic lineage in 2 (13%) cases. Ringed sideroblasts were documented in one case.
Cytogenetic results at the time of the diagnosis were available for all 16 cases (Tables 2 and 7 ). The karyotype was complex in 7 of 16 (44%) cases; unbalanced cytogenetic abnormalities were observed predominantly in the context of a complex karyotype. Exceptions included two cases of del(20)(q11.2), one of which was associated with monosomy 7. Trisomy 8 was observed exclusively in the context of complex cytogenetic abnormalities.
Survival and Clinical Follow-Up
Given that the patients did not receive uniform therapy, we only compared the overall survival of the four subgroups. Patients with acute erythroid leukemia had a better overall survival than patients with acute myeloid leukemia with myelodysplasia-related changes or therapy-related acute myeloid leukemia (P=0.01 and P<0.0001, respectively; Table 8 , Figure 2).

Overall survival of patients by diagnostic subgroups. Overall survival of patients with acute myeloid leukemia or myelodysplastic syndrome with erythroid predominance. (a) Overall survival of patients in four diagnostic subgroups. (b) Comparison of overall survival between acute erythroid leukemia and acute myeloid leukemia with myelodysplasia-related changes and (P=0.005). (c) Comparison of overall survival between acute erythroid leukemia and therapy-related acute myeloid leukemia (P<0.0001). (d) Comparison of overall survival between acute erythroid leukemia and refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia.
We also analyzed the influence of different variables on overall survival. The only statistically significant variable that adversely affected survival in the acute erythroid leukemia (P=0.007) and acute myeloid leukemia with myelodysplasia-related changes (P=0.004) subgroups was the presence of a complex karyotype (Table 9 , Figure 3). We also compared overall survival of patients with acute erythroid leukemia without a complex karyotype to that of patients in other subgroups. We found an even more pronounced survival advantage for acute erythroid leukemia patients without complex karyotype when compared with other subgroups (Table 10 , Figure 4). In the acute erythroid leukemia subgroup, a comparison of abnormal karyotype (including simple and complex abnormalities) versus diploid karyotype did not correlate with survival. In the therapy-related acute myeloid leukemia and refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia subgroups, there was no statistically significant difference in survival based on presence/absence of a complex karyotype. In the therapy-related acute myeloid leukemia subgroup, the type of previous malignancy (hematolymphoid versus solid tumor) did not correlate with survival (Figure 5).

Effect of a complex karyotype on overall survival of patients with acute myeloid leukemia or myelodysplastic syndrome with erythroid predominance. (a) Comparison of overall survival in acute erythroid leukemia patients (P=0.007). (b) Comparison of overall survival in patients with acute myeloid leukemia with myelodysplasia-related changes (P=0.004). (c) Comparison of overall survival in therapy-related acute myeloid leukemia patients (P=0.524). (d) Comparison of overall survival in patients with refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia (P=0.072).

Comparison of survival of patients with acute erythroid leukemia without a complex karyotype versus other patients with acute myeloid leukemia or myelodysplastic syndrome with erythroid predominance.

Survival of patients with therapy-related acute myeloid leukemia and its relationship to the type of previous neoplasm.
Discussion
The concept and definition of acute erythroid leukemia has evolved substantially over the years. The most recent update of the World Health Organization classification further specifies the criteria of acute erythroid leukemia, as well as criteria for the new entity, acute myeloid leukemia with myelodysplasia-related changes.17 The current World Health Organization classification also recognizes therapy-related acute myeloid leukemia.18 A major goal of this study was to study the effect of the World Health Organization classification criteria on cases that were interpreted previously as acute erythroid/myeloid leukemia, erythroleukemia or acute myeloid leukemia of M6 type using previous classification systems at our institution over the past 17 years. Application of the current World Health Organization criteria lead to re-classification of a considerable number of cases originally diagnosed as erythroleukemia as either acute myeloid leukemia with myelodysplasia-related changes or therapy-related acute myeloid leukemia. A subset of patients treated with erythropoietin was also reclassified as a myelodysplastic syndrome.
We believe the cytogenetic data in the subgroup of acute erythroid leukemia cases is noteworthy because the data suggest that acute erythroid leukemia as currently defined remains somewhat heterogeneous. The 20 patients of acute erythroid leukemia in this study can be divided into two groups: 15 patients with diploid cytogenetics or non-complex cytogenetic abnormalities and 5 patients with complex karyotypes. The first group of 15 patients is of interest because their clinical course was relatively indolent. The median blast count in this group was 16% of all nucleated cells. The diagnosis of acute erythroid leukemia was established on the basis of erythroid predominance with blasts representing 20% or more of all nonerythroid cells in the bone marrow. However, perhaps the lower 20% cutoff used by the World Health Organization classification has had the effect of ‘overcalling’ some cases of myelodysplastic syndrome as acute erythroid leukemia when there is erythroid predominance. Using the French–American–British classification cutoff for acute myeloid leukemia of 30%, only one case in the acute erythroid leukemia group would have met the criteria for acute erythroid leukemia. This issue has been raised previously by Selby et al22 and has important implications for therapy of these patients.
A second group of acute erythroid leukemia cases, five patients with complex karyotypes, probably fits within the spectrum of acute myeloid leukemia with myelodysplasia-related changes. This opinion is based on the presence of multilineage dysplasia in the aspirate smears of four patients and the cytogenetic abnormalities, which were predominantly unbalanced and commonly associated with myelodysplastic syndrome as has been reported in the literature.23, 24 The presence of complex cytogenetics in these five patients also correlated with an adverse effect on overall survival. However, in all five cases bone marrow blasts were <20% (range, 7–15%) and therefore these cases did not meet the World Health Organization criteria for acute myeloid leukemia with myelodysplasia-related changes. The 20% cutoff, of course, is an arbitrary number and perhaps a complex karyotype should be used to exclude cases from the category of acute erythroid leukemia in the future versions of the World Health Organization classification.
The patients with acute erythroid leukemia have some distinctive clinical and biologic features. Patients in this group were younger, their disease had a high frequency of diploid cytogenetics associated with less pronounced dysplasia in hematopoietic elements, and they seemed to follow a more favorable clinical course than patients in the other subgroups. Patients in this subgroup had the highest frequency of relapse (11/20, 55%). This high relapse rate may be a reflection of responsiveness to chemotherapy. Erythroid predominance was not a feature at relapse in 8 of 11 (73%) patients. In spite of the relapse rate, acute erythroid leukemia patients had a statistically better overall survival than patients in the acute myeloid leukemia with myelodysplasia-related changes and therapy-related acute myeloid leukemia groups. The acute erythroid leukemia subgroup was the only group to show fms-like tyrosine kinase 3 gene abnormalities, and these abnormalities were observed in patients with either diploid or non-complex cytogenetic abnormalities. The high percentage of diploid cytogenetics in the acute erythroid leukemia group suggests that there are cryptic cytogenetic or molecular abnormalities yet to be discovered, which have a role in pathogenesis.
In contrast, patients with therapy-related acute myeloid leukemia and erythroid predominance had the highest percentage of complex cytogenetic abnormalities and the worst overall survival among the four subgroups. The cytogenetic abnormalities in therapy-related acute myeloid leukemia were similar to those observed in the acute myeloid leukemia with myelodysplasia-related changes and refractory anemia with excess blasts and recent/concurrent history of erythropoietin treatment for anemia groups. The association with complex cytogenetics probably contributes the most to their poor outcome. Overall survival of therapy-related acute myeloid leukemia patients did not correlate with the type of the preceding neoplasm (hematolymphoid versus solid tumor).
We acknowledge that the category of refractory anemia with excess blasts and preceding/concurrent history of erythropoietin therapy used in this study is heterogeneous and not well characterized. We decided to segregate this group because erythropoietin therapy, by increasing erythroid precursors in the bone marrow, complicates the determination of blast counts. As a result of the iatrogenic elevation of the erythroid count, a case of ‘true’ myelodysplastic syndrome may be overcalled as acute erythroid leukemia after the substraction of erythroid precursors before the blast count. Conversely, a case of ‘true’ acute myeloid leukemia with myelodysplasia-related changes may be undercalled because of the increased erythroid precursors being included as a denominator in the blast count. In spite of the poor diagnostic definition of the diseases in this category, it is of interest that the median survival was relative poor, 17 months. This may be explained by the high frequency of bi- or trilineage dysplasia and abnormal cytogenetic findings in this group. Fourteen (88%) patients had bi- or trilineage and 13 (81.3%) patients had cytogenetic abnormalities including 11 patients with a complex karyotype. Abnormalities of chromosomes 5 and 7 were also common in patients with cytogenetic abnormalities. In aggregate, these data suggest erythroid predominance combined with obvious morphologic evidence of dysplasia and/or abnormal cytogenetic findings are features of a disease with a poor prognosis. Similar findings are reported by Wang et al.25 Perhaps the exact blast count or erythroid precursor counts are not critical in the design of a treatment plan for these patients.
In conclusion, in the past leukemias of erythroid lineage designated previously as erythroleukemia, acute myeloid leukemia M6 type, or acute erythroid/myeloid leukemia were thought to represent 4–5% of all cases of acute myeloid leukemia and to have a poor prognosis. In this study, through rigorous application of the current World Health Organization criteria, we have shown that acute erythroid leukemia is a much more infrequent disease with an overall favorable outcome when compared with acute myeloid leukemia with myelodysplasia-related changes and therapy-related acute myeloid leukemia presenting with erythroid predominance. Furthermore, the category of acute erythroid leukemia as defined in the World Health Organization classification is still heterogeneous. Approximately 75% of cases have diploid or non-complex cytogenetics and an indolent clinical course. The blast count in this group is relatively low, <20% of total nucleated cells in many cases, and perhaps the diagnosis of acute erythroid leukemia in this subgroup leads to a more aggressive therapy than is needed. By contrast, approximately 25% of acute erythroid leukemia cases are associated with complex karyotypes and a more aggressive clinical course, and seem to be closely related to acute myeloid leukemia with myelodysplasia-related changes. The results of this study suggest that the World Health Organization criteria for acute erythroid leukemia may need to be further refined in the next update of this classification.
References
Hetzel P, Gee TS . A new observation in the clinical spectrums of erythroleukemia. A report of 46 cases. Am J Med 1978;64:765–772.
Dameshek W, Baldini M . The Di Guglielmo syndrome. Blood 1958;13:192–194.
Fouillard L, Labopin M, Gorin NC, et al. Hematopoietic stem cell transplantation for de novo erythroleukemia: a study of the European Group for Blood and Marrow Transplantation (EBMT). Blood 2002;100:3135–3140.
Dameshek W . The DiGuglielmo syndrome revisited. Blood 1969;34:567–572.
Bain BJ . Di Guglielmo and his syndromes. Br J Haematol 2003;120:939–943.
Martin WJ, Bayrd ED . Erythroleukemia, with special emphasis on the acute or incomplete variety; report of five cases. Blood 1954;9:321–339.
Dreyfus B, Rochant H, Sultan C . Refractory anemias: acquired enzymopathies of hematopoietic stem cells. Nouv Rev Fr Hematol 1969;9:65–86.
Kiossoglou KA, Mitus WJ, Dameshek W . Chromosomal aberrations in acute leukemia. Blood 1965;26:610–641.
Dyment PG, Melnyk J, Brubaker CA . A cytogenetic study of acute erythroleukemia in children. Blood 1968;32:997–1002.
Heath Jr CW, Bennett JM, Whang-Peng J, et al., Wiernick PH Cytogenetic findings in erythroleukemia. Blood 1969;33:453–467.
Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976;33:451–458.
Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 1985;103:620–625.
Brunning RD, Matutes E, Flandrin G, et al. Acute myeloid leukaemia not otherwise catogorised In: Jaffe ES (ed). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, France, 2001, pp 91.
Arber DA, Brunning RD, Orazi A, et al. Acute myeloid leukaemia, not otherwise specified In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. IARC Press: Lyon, France, 2008, pp 130.
Goasguen JE, Matsuo T, Cox C, et al. Evaluation of the dysmyelopoiesis in 336 patients with de novo acute myeloid leukemia: major importance of dysgranulopoiesis for remission and survival. Leukemia 1992;6:520–525.
Oyarzo MP, Lin P, Glassman A, et al. Acute myeloid leukemia with t(6;9)(p23;q34) is associated with dysplasia and a high frequency of flt3 gene mutations. Am J Clin Pathol 2004;122:348–358.
Arber DA, Brunning RD, Orazi A, et al. Acute myeloid leukaemia with myelodysplasia-related changes In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, France, 2008, pp 124.
Vardiman JW, Arber DA, Brunning RD, et al. Therapy-related myeloid neoplasms In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, France, 2008, pp 127.
Onciu M, Schlette E, Medeiros LJ, et al. Cytogenetic findings in mantle cell lymphoma cases with a high level of peripheral blood involvement have a distinct pattern of abnormalities. Am J Clin Pathol 2001;116:886–892.
Chen W, Konoplev S, Medeiros LJ, et al. Cuplike nuclei (prominent nuclear invaginations) in acute myeloid leukemia are highly associated with FLT3 internal tandem duplication and NPM1 mutation. Cancer 2009;115:5481–5489.
Gustafson SA, Lin P, Chen SS, et al. Therapy-related acute myeloid leukemia with t(8;21) (q22;q22) shares many features with de novo acute myeloid leukemia with t(8;21)(q22;q22) but does not have a favorable outcome. Am J Clin Pathol 2009;131:647–655.
Selby DM, Valdez R, Schnitzer B, et al. Diagnostic criteria for acute erythroleukemia. Blood 2003;101:2895–2896.
Bernasconi P . Molecular pathways in myelodysplastic syndromes and acute myeloid leukemia: relationships and distinctions-a review. Br J Haematol 2008;142:695–708.
Scholl S, Fricke HJ, Sayer HG, et al. Clinical implications of molecular genetic aberrations in acute myeloid leukemia. J Cancer Res Clin Oncol 2009;135:491–505.
Wang SA, Tang G, Fadare O, et al. Erythroid-predominant myelodysplastic syndromes: enumeration of blasts from nonerythroid rather than total marrow cells provides superior risk stratification. Mod Pathol 2008;21:1394–1402.
Acknowledgements
We thank the following pathologists at our institution who signed out individual cases included in this study: Cheryl Hirsh-Ginsberg, MD, Hesham M Amin, MD, Yang O Huh, MD, Jeffrey L Jorgensen, MD, Sergej N Konoplev, MD, PhD, Pei Lin, MD, Ellen Schlette, MD, Francisco Vega-Vasquez, MD, PhD, Sa Wang, MD, C Cameron Yin, MD, PhD, Lynne V Abruzzo, MD, PhD, Dan Jones, MD, PhD and M James You, MD, PhD. We thank Beverly Jean Johnson, MT, Steven Reyes, MT, Rosalina Sarinas, MT and Esther Moreno, MT for their excellent technical help; Kathryn Hale for her editorial suggestions and La Kisha Rodgers for secretarial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kasyan, A., Medeiros, L., Zuo, Z. et al. Acute erythroid leukemia as defined in the World Health Organization classification is a rare and pathogenetically heterogeneous disease. Mod Pathol 23, 1113–1126 (2010). https://doi.org/10.1038/modpathol.2010.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.96
Keywords
This article is cited by
-
An analysis of 97 previously diagnosed de novo adult acute erythroid leukemia patients following the 2016 revision to World Health Organization classification
BMC Cancer (2017)
-
Acute erythroid leukemia with <20% bone marrow blasts is clinically and biologically similar to myelodysplastic syndrome with excess blasts
Modern Pathology (2016)
-
Dysplasia Has A Differential Diagnosis: Distinguishing Genuine Myelodysplastic Syndromes (MDS) From Mimics, Imitators, Copycats and Impostors
Current Hematologic Malignancy Reports (2012)
