
Abstract
TH17 cells are major drivers of inflammation and involved in several autoimmune diseases. Tissue inflammation is a beneficial host response to infection, but it can also contribute to autoimmunity. The crosstalk between a tissue and the immune system during an inflammatory response is key for preserving tissue integrity and restoring physiological processes. However, how the inflamed tissue regulates the magnitude of an immune response by controlling pro-inflammatory T cells is not well characterized so far. Here we show that TH17 cells accumulating in the small intestine upon inflammation express the IL-33 receptor (ST2) and intestinal epithelial cells (IEC) are the main source of the alarmin interleukin-33 (IL-33). We show that pro-inflammatory TH17 cells acquire a regulatory phenotype with immunosuppressive properties in response to IL-33. Absence of ST2 signaling promotes the secretion of pro-inflammatory cytokines by TH17 cells and dampens the secretion of IL-10. Our results provide new insights into the mechanisms by which IEC, via IL-33/ST2 axis, may control pro-inflammatory TH17 cells in the small intestine to sustain homeostasis.
Similar content being viewed by others


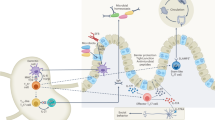
INTRODUCTION
The TH17 cell lineage represents an effector T-cell subset particularly enriched in the small intestinal lamina propria, where it protects the host from bacterial infections.1 However, when their regulation fails, TH17 cells also have the potential of becoming key drivers of several autoimmune disorders like multiple sclerosis2 and inflammatory bowel disease.3 Given that TH17 cells have emerged as heterogeneous and flexible cells, highlighting how cell plasticity plays a central role in keeping the balance between pathogenicity and tolerance,4 it is of crucial importance to understand how the diverse functions of this cell subset are regulated.
In vivo animal models of tolerance induction, such as anti-CD3 treatment, have identified the small intestine (SI) as a major regulatory organ not only for local, but also for systemic immune function.5 Anti-CD3 treatment leads to two well characterized and distinct phases in the subsequent immune response. The first phase (day 1) results in a systemic immune stimulation provoked by a strong cytokine release that results in flu-like symptoms and in the disappearance of T cells from the periphery,6 together with T-cell dependent small intestinal injury.7 Nevertheless, the cytokine release is only transient and self-limiting and it resolves with the complete restoration of the intestinal tissue and systemic homeostasis8 by the fourth or fifth day of treatment, corresponding to the second phase, the resolution of inflammation. Resolution of inflammation leads to active systemic immunological tolerance mediated by regulatory T cells. In the anti-CD3 treatment model of tolerance induction, as well as in infection models such as sepsis and during influenza A viral infection (H1N1), pro-inflammatory TH17 cells are recruited to the SI via the CCR6/CCL20 axis.5 In the SI, TH17 cells transdifferentiate into regulatory T cells, which are able to effectively suppress proliferation of effector T cells both in vitro and in vivo.5, 9, 10 The local tissue response upon injury and the tissue-specific accumulation of TH17 cells makes anti-CD3 treatment a good model to study TH17 cells in inflamed tissue, as well as the subsequent modulation of the ongoing T-cell response. In consideration of evidence pointing to the intestine as a crucial immune organ, where tolerance is strictly and efficiently maintained to avoid unwanted immune responses to self-, dietary, or commensal antigens,11, 12 it is of great interest to comprehend tissue-specific mechanisms, by which the gut is capable of inducing tolerance in TH17 cells.
Interleukin-33 (IL-33) is a member of the IL-1 cytokine family,13 mainly expressed by endothelial and epithelial cells at mucosal interfaces14 and known to function as an alarmin, a molecule rapidly indicating tissue damage upon injury or infection.15 Although IL-33 has been linked to various inflammatory processes and autoimmune diseases,16 recent studies have described the role of IL-33 in tissue regulation and repair, especially in the intestine.17 IL-33 signals through the ST2 receptor, expressed by multiple non-hematopoietic and hematopoietic cells. The wide distribution of ST2 and the interplay between its constitutive and induced expression point to a key role of the IL-33/ST2 axis in both inflammation and homeostasis, affecting innate as well as adaptive immune responses. A deeper knowledge of the tissue- and cell-specific mechanisms by which the IL-33/ST2 axis governs both processes in the intestine remains an important area for exploration.
In this study, we found that epithelial cells in the proximal part of the SI are the main source of IL-33 upon inflammation. We describe for the first time ST2 expression in TH17 cells in vivo and we show that in both mouse and human TH17 cells, IL-33 promotes a reduced expression of pro-inflammatory genes, including Tbx21, Ifng, and Csf2, and an increase in Il10, an anti-inflammatory cytokine that has non-redundant roles in limiting inflammation in the intestine18 and in controlling previously committed pathogenic TH17 cells.5, 9, 19 Our in vivo studies performed with St2−/− mice suggest a novel role for the IL-33/ST2 axis not only in limiting the expansion of TH17 cells in the SI during inflammation, but also in dampening the inflammatory capacity of these cells. In addition, in vivo blocking of ST2 leads to a reduction in the expression of IL-10 by TH17 cells in the SI. These findings, together with the suppression of the proliferation of responder T cells by IL-33-stimulated TH17 cells in vitro, suggest that TH17 cells acquire an immunosuppressive phenotype in response to IL-33. Our results provide new insights into the mechanisms by which intestinal epithelial cells (IEC), via IL-33, may control pathogenic TH17 cell responses in the SI, to maintain homeostasis.
RESULTS
Epithelial cells in the proximal part of the SI are the major source of the alarmin IL-33 upon inflammation
To identify tissue-derived signals involved in the control of pathogenic TH17 cells in the SI, we first performed a gene expression analysis of non-hematopoietic cells. The results of the microarray showed a significant upregulation of Il33 in the CD45− population from the inflamed SI of anti-CD3-treated mice, compared with the CD45− population from mice in steady state (Supplementary Figure S1a). Il33 mRNA levels were upregulated in the CD45−-cell fraction of treated animals compared with CD45− cells from untreated mice, confirming the transcriptome data (Supplementary Figure S1b). To further decipher the specific cellular source of IL-33 in the SI, we performed immunofluorescence staining of tissue sections. Extensive IL-33 staining was detected in EpCAM+epithelial cells located in the apical part of the villi in the duodenum of treated mice (Figure 1a). By contrast, IL-33 expression in steady state was only detected in isolated PECAM+ endothelial cells located in the crypt region (Figure 1a). Histological analysis of peripheral lymphoid tissues revealed only few cells expressing IL-33 in the spleen and mesenteric lymph nodes and no differences were observed upon anti-CD3 treatment (Figure 1b). Thus, we could confirm that IL-33 is a local, tissue-specific signal induced upon inflammation. Endothelial cells represent the basal source of IL-33 under homeostatic conditions, while IECs represent the major source of the alarmin IL-33 upon inflammation. To further analyze the spatial and temporal distribution of IL-33 in various parts of the SI, we performed a time-course analysis of the transcript during anti-CD3 treatment (Supplementary Figure S1c). In line with our previous findings, Il33 levels were 450-fold upregulated at day 1, decreasing rapidly afterwards. Il33 was mainly restricted to the duodenum and, to a lesser extent, to the jejunum (Figure 1c). We confirmed the upregulation of Il33 in the proximal part of the SI from anti-CD3-treated mice by immunofluorescence microscopy. Notably, IL-33 decreased toward the distal parts in a gradient-like fashion, with only marginal expression in the ileum and non-detectable expression in the colon (Figure 1d). To explore in depth the kinetics of Il33 specifically in the duodenum, where its highest levels were observed, we analyzed the tissue at 4 and 24 h after each injection. Indeed, a peak of Il33 was observed 4 h after the first injection (day 1), after which the levels decreased to a value sixfold less (Figure 1e). However, the transcript was upregulated within 4 h after every additional injection, but only reaching a twofold increase. The acute and time-limited expression of Il33 in the inflamed SI emphasized its role as an alarmin. The kinetics was confirmed by immunofluorescence staining of the duodenum (Figure 1f), where histological analysis also showed that, upon anti-CD3 treatment, upregulation of IEC-derived IL-33 (green) was concurrent with the recruitment of TH17 cells (red) in the lamina propria (LP). Importantly, anatomical localization of IL-33 corresponded to distinct parts of the intestine where TH17 cells accumulate.5 On the basis of these data, we aimed to determine whether accumulating SI TH17 cells may respond to IL-33.

IL-33 is upregulated in intestinal epithelial cells (IEC) in the proximal part of the small intestine upon CD3-specific antibody injection. C57BL/6 (a–d) or IL17A-eGFPxFoxp3-mRFP reporter (e–f) mice were injected i.p. with anti-CD3 at days 1, 3, and 5. Four hours after each injection, the different parts of the intestine were isolated, as well as the spleen and mesenteric lymph nodes (MLN) (control tissues). (a) Histological analysis of IL-33 (green), EpCAM (red), and PECAM (blue) in the duodenum is shown at steady state and after the first injection of anti-CD3 antibody. (b) Histological analysis of IL-33 (green) and Hoechst (white, nuclear counterstaining) in the spleen and MLN is shown at steady state and after the first injection of anti-CD3 antibody. (c) Quantitative PCR analysis (qPCR) of Il33 mRNA was performed for each tissue at every time point. (d) Immunohistochemistry (IHC) analysis of IL-33 (green), EpCAM (red), and PECAM (blue) in the different parts of the SI in steady state and upon anti-CD3 injection. (e) Duodenal Il33 mRNA was measured by qPCR 4 and 24 h after each injection. (f) IHC of the duodenum at the indicated time points showing the localization of IL-33 (green), IL-17A (red, GFP), CD11c (blue), and Hoechst (white, nuclear counterstaining). Red arrows indicate anti-CD3 injections. qPCR data are presented as mean±s.e.m. and normalized to the values in steady state. Data are representative of ≥3 independent experiments (n=4 animals per group).
TH17 cells show inducible ST2 expression
IL-33 mediates its function via ST2, a plasma membrane receptor that has been described to be expressed in various T-cell subsets including TH2,20 TH1,21 and Tregs.22 However, it is not known whether TH17 cells express ST2. Therefore, we analyzed ST2 expression by flow cytometry in the spleen and the SI of IL17A-eGFPxFoxp3-mRFP double reporter mice at steady state and during anti-CD3 treatment (gating strategies and control staining for ST2 shown in Supplementary Figure S2a and b, respectively). We could not find ST2+ TH17 cells in the SI of untreated mice (Figure 2a and c). However, increasing frequencies of TH17 cells were found in the ST2+ T-cell fraction upon repetitive CD3-specific antibody injections in the SI, reaching ∼20% during resolution of inflammation (day 5) (Figure 2a and c). Frequencies of Tregs in the intestinal ST2+ T-cell pool decreased during anti-CD3 treatment from 60% at steady state to 20% at day 5 (Figure 2a and c). Similarly, ST2+ TH17 cells were absent in the spleen at steady state, but appeared upon anti-CD3 treatment and their frequency increased, reaching ∼15% of the total ST2+ T-cell population during the resolution of inflammation (Figure 2b and c). Frequencies of ST2+ Tregs in this tissue decreased from ∼55 to 35% during anti-CD3 treatment (Figure 2b and c).

TH17 cells express ST2 in vivo upon anti-CD3 treatment. IL17A-eGFPxFoxp3-mRFP reporter mice were injected i.p. with anti-CD3 at day 1, 3 and 5. Flow cytometry analysis of TH17 cells (gated as Foxp3−IL-17A+) and Tregs (gated as IL-17A−Foxp3+) contained in the ST2+ T-cell population at steady state and during anti-CD3 treatment from the SI (a) and the spleen (b). (c) Frequencies of ST2+ TH17 and ST2+ Tregs at steady state and during anti-CD3 treatment in both tissues, gated as in a–b. (d) Flow cytometry analysis of ST2 and CCR6 in splenic TH17 and Tregs after anti-CD3 treatment (day 5). (e) Comparison of the mean fluorescence intensity of ST2 in TH17 cells during anti-CD3 treatment in spleen and SI. Data are shown as mean±s.e.m. and are representative of two independent experiments (n=5 animals per group). *P<0,05, ****P<0,0001 as calculated by two-way Anova with Bonferroni post-test. ns, not significant. Backgating strategy and controls for ST2 staining shown in Supplementary Figure S2a and b, respectively.
Splenic TH17 cells express CCR6, a chemokine receptor known for playing a role on directing TH17 cells toward the SI, where high amounts of CCL20, the ligand of CCR6, are released during anti-CD3 treatment.5 Indeed, ∼90% of splenic TH17 cells expressed CCR6 during the resolution of inflammation (day 5) and, interestingly, ST2 expression in this subset was largely restricted to CCR6-expressing cells, while ST2 expression in Tregs was mainly restricted to the CCR6− population (Figure 2d). We also compared the expression of the gut homing receptor CCR6 between ST2− and ST2+ TH17 cells in the spleen and the SI during anti-CD3 treatment. While ST2+ TH17 cells expressed high levels of CCR6 (∼60%) in both tissues during resolution of inflammation, ST2− TH17 expressed high levels of CCR6 only in the spleen, but reduced levels (∼15%) in the SI (Supplementary Figure S3).
Although frequencies of TH17 cells were higher in the intestinal ST2+ T-cell pool than in the spleen, the analysis of the mean fluorescence intensity of ST2 in TH17 cells revealed a reduction in the SI, indicating that TH17 in this tissue expressed reduced levels of ST2 at the single cell level, compared with the spleen (Figure 2e). Together, these data demonstrate an inducible expression of ST2 in TH17 cells upon anti-CD3 treatment.
Absence of ST2-mediated signaling favors the expansion of pro-inflammatory TH17 cells in the SI upon inflammation
In light of the finding that TH17 cells express ST2 in vivo, we next aimed to assess the role that the IL-33/ST2 signaling pathway plays in these cells. For that, TH17 responses upon anti-CD3 treatment were compared between St2−/− mice and control littermates (Figure 3a; gating strategies shown in Supplementary Figure S2a). Because we found the strongest upregulation of IL-33 during the acute inflammation phase (day 1) (Figure 1c and f), we expected the greatest difference between St2−/− and control mice at this time point. In the absence of ST2 signaling, frequencies of RORγt+, Foxp3+, and RORγt+/Foxp3+ cells were significantly increased in the spleen, while frequencies of cytokine-expressing T cells did not change between the two cohorts (Figure 3b). We observed a selective enrichment in the SI of St2−/− mice, both in frequencies and total numbers, of RORγt-expressing cells (Figure 3c and d). By contrast, frequencies of intestinal Foxp3- expressing cells and Foxp3/RORγt- double expressing cells were unaffected by ST2 deficiency (Figure 3c). Interestingly, elevated frequencies and total numbers of IL-17A+ T cells were also found in the SI in the absence of St2, as well as higher frequencies of IFN-γ/IL-17A- and TNF-α/IL-17A- double expressing cells (Figure 3c and d), suggesting that St2 deficiency specifically enhances the pro-inflammatory capacity of TH17 cells in our settings. However, no significant differences were observed in IFN-γ or TNF-α single production by T cells (Figure 3c). The hypothesis of an exacerbated TH17 pro-inflammatory response in the absence of ST2 signaling was further supported by the gene expression analysis of in vitro differentiated St2−/− TH17 cells, which showed enhanced expression of Rorc, Il17a, and Ifng when compared with WT TH17 cells (Figure 3e).

St2 −/− mice show a specific increase of pathogenic TH17 cells in the SI upon anti-CD3 injection. (a) St2 deficient (St2−/−) and wild-type C57BL/6 (wt) mice were injected i.p. with anti-CD3 and T cells were isolated from SI and spleen 4 h after injection to be characterized by flow cytometry. A representative FACS dot plot for each group (n=4) and the associated frequencies of TF-(RORγt and Foxp3) and cytokine-(IL-17A, IFN-γ, TNF-α) expressing T cells isolated from the spleen (b) or the SI (c) are shown. (d) Absolut numbers of CD45+CD4+TCRβ+RORγt+ or IL-17A+ T cells in the SI. Results are shown as mean±s.e.m. and are representative of two independent experiments.* P<0,05, **P<0,01, ***P<0,005 as calculated by unpaired Student’s t-test. ns, not significant. Backgating strategy shown in Supplementary Figure S2a. (e) Mouse naive sorted CD4+CD25−CD62L+CD44− T cells from St2−/− or wt mice were stimulated in vitro with anti-CD3/CD28 and grown under TH17 conditions for 5 days. Expression of Rorc, Il17a, and Ifng in St2−/− TH17 cells was analyzed by qPCR and compared with wt TH17 cells (dotted line). Data are shown as mean±s.d. and are representative of three independent experiments.
The analysis at day 3 after treatment showed, as expected, milder differences between groups (Supplementary Figure S4a). At day 5, no differences were detected (Supplementary Figure S4b). We also compared T-cell populations under homeostatic conditions and St2−/− mice and control littermates showed similar levels of T-cell-derived cytokines in the spleen and the SI (Supplementary Figure S4c), in line with the absence of any immune phenotype observed in these mice and confirming what has already been reported for the St2−/− mouse strain.23 Hence, the differences observed in the SI TH17 population in St2−/− mice at day 1 were induced by the treatment, suggesting a link between IL-33/ST2 axis and the ongoing acute inflammatory response. Together, our results show that the IL-33/ST2 axis specifically limits the expansion of pathogenic TH17 cells in the SI upon inflammation. The exacerbated intestinal TH17 response observed in St2−/− mice during the acute inflammatory phase also led to clinical manifestations. Mice lacking St2 showed greater body weight loss upon anti-CD3 treatment (Supplementary Figure S5a), as well as a higher inflammation score in the duodenum (Supplementary Figure S5b). This was associated with an enhanced inflammatory cytokine milieu, as depicted by an upregulation of the pro-inflammatory genes Il17a, Ifng, and Tnf, together with a downregulation of the anti-inflammatory gene Il10 (Supplementary Figure S5c).
It has already been reported by us that during resolution of inflammation, TH17 cells located in the LP display an immunosuppressive phenotype characterized by their ability to produce IL-10. Consequently, they are able to effectively suppress the proliferation of naive T cells in vitro and ameliorate the clinical course of experimental autoimmune encephalomyelitis (EAE), demonstrating their in vivo regulatory function.5, 9, 10 To analyze whether ST2 signaling in TH17 cells may also have an impact on IL-10 induction, we followed two different strategies. On the one hand, we administered a blocking antibody against ST2 in vivo24, 25 prior to anti-CD3 injections. On the other hand, we used IL-10-GFP reporter mice with genetically deleted St2 (IL-10-GFP.T1ST2KO mice). In both cases, we analyzed intestinal TH17 cells during resolution of the inflammation, the phase in which the highest levels of T-cell-derived IL-10 were expected (Figure 4a). The frequency of intestinal RORγt+ T cells was found increased in the anti-ST2-treated group compared with the group treated with control antibodies (Figure 4b). These results were similar to our observations in the IL-10-GFP.T1ST2KO mice (Figure 4c), pointing to an effective in vivo blockade of the receptor. Interestingly, the intestinal TH17 population in the anti-ST2-treated group not only showed increased frequencies of IL-17A producers, but also reduced frequencies of IL-10+ TH17 cells (Figure 4d). Analysis of IL-10-GFP.T1ST2KO mice showed similar results (Figure 4e), suggesting that absence of ST2 signaling leads to a reduction of IL-10 secretion by TH17 cells, supporting their pro-inflammatory phenotype.

Absence of ST2 leads to increased frequencies of intestinal TH17 cells that are characterized by a limited production of IL-10. (a) IL-10-GFP reporter mice or IL-10-GFP.T1ST2KO mice were injected i.p. with anti-CD3 antibodies and T cells were isolated from the SI after the second injection for subsequent FACS analysis. IL-10-GFP reporter mice were injected i.p with anti-ST2 or isotype control (3–5 animal per group) 30 min prior to anti-CD3 injections. A representative dot plot for each animal group and associated frequencies of TH17 cells (gated as CD45+CD4+TCRβ+RORγt+ T cells) are shown in b for the in vivo blocking of ST2 and in c for IL-10-GFP.T1ST2KO mice. A representative dot plot of IL-17A and IL-10 expression in TH17 cells (gated as in b) for each animal group and associated frequencies of cytokine expression among TH17 cells are shown in d for the in vivo blocking of ST2 and in e for IL-10-GFP.T1ST2KO mice. Data are shown as mean±s.e.m. of at least two independent experiments. *P<0,05, **P<0,01, ***P<0,005, ****P<0,001 as calculated by unpaired Student’s t-test. Backgating strategy shown in Supplementary Figure S2a.
IL-33 induces changes in the gene expression profile of mouse and human TH17 cells in favor of an immunosuppressive phenotype
Our next goal was to further elucidate specific and direct effects of IL-33 on TH17 cells. We first assessed the expression of ST2 in naive T cells, known to respond to IL-33 in vitro,26 and in vitro differentiated TH17 cells and Tregs (Figure 5a). Although in vitro differentiated murine TH17 cells expressed low levels of ST2, they responded to IL-33 stimulation. Il10 expression was induced fourfold in the presence of IL-33 and the increase in Il10 expression was accompanied by a reduction in the levels of Tbx21, Ifng, and Csf2 (Figure 5b). Importantly, human TH17 cells responded similarly to IL-33 stimulation. IL10 was upregulated in the presence of IL-33, while TBX21 levels were remarkably reduced (Figure 5c). On the protein level, IL-10 production in human TH17 cells was also induced by IL-33 (Figure 5d) compared with untreated cells and to cells treated with IL-1β, a reported inhibitor of IL-10 in human TH17 cells.27 As expected, the maximum of IL-10 production was found in TH17 cells treated with IL-27, a cytokine known to strongly induce IL-10.28 Notably, IL-33 also dampened the production of the pro-inflammatory cytokine granulocyte macrophage-colony stimulating factor (GM-CSF) by human TH17 cells (Figure 5d). Together, these data indicate that IL-33 is capable of inducing changes in the genetic profile of murine and human TH17 cells in favor of a reduced pathogenic phenotype and promotes the expression of the anti-inflammatory cytokine IL-10. This was also supported by our finding that Ahr,10 Ikzf3,19 Jun, and Irf4,29 all known to be IL-10-related genes, were found to be upregulated by IL-33 in TH17 cells, together with Il10 (Figure 5e). Expression of Ahr was also found to positively correlate with Il10 expression in TH17 cell cultures (Supplementary Figure S6), suggesting that the transcription factor Ahr may be involved in the induction of Il10 downstream of IL-33/ST2 signaling. Although Maf is a reported inducer of Il10 in TH17 cells,30 it was not upregulated upon IL-33 signaling (Figure 5e). Nevertheless, expression of Maf was found to correlate with Il10 expression in TH17 cell cultures (Supplementary Figure S6), indicating a link between both transcripts, but not with IL-33.

Mouse and human TH17 cells respond to IL-33 in vitro. (a) Mouse naive sorted CD4+CD25−CD62L+CD44− T cells were stimulated in vitro with anti-CD3/CD28 and grown under TH17 or Treg conditions for 5 days. ST2 expression was analyzed by qPCR and compared with naive cells (mean±s.d. of three independent experiments). (b) Mouse TH17 cells were treated with 20 ng ml−1 IL-33 during the differentiation process (as in a). Expression levels of Il10, Tbx21, Ifng, and Csf2 were quantified by qPCR (dotted line at 1 indicates the value of control untreated TH17 cells). Data are presented as mean±s.e.m. of ≥3 independent experiments. (c) Fresh ex vivo sorted human CCR6+CCR4+CXCR3–CD45RA–CD25–CD8– TH17 cells were stimulated with CD3 and CD28 mAbs for 48 h and expanded for another 3 days in the presence of 20 ng ml−1 IL-33. IL10 and TBX21 expression levels were quantified by qPCR (dotted line at 1 indicates value of non-treated TH17 cells). Human TH17 cells were also stimulated with PMA/Ionomycin for 5 h to detect intracellular levels of IL-17A, IL-10 and GM-CSF by FACS. One representative dot plot out of three independent experiments of flow cytometry analysis of IL-10 and GM-CSF assessed on TH17 cells (gated on IL17A+ CD4+ T cells) is shown in d. (e) Mouse TH17 cells were treated with 20 ng ml−1 IL-33 during the differentiation process (as in a) and the expression levels of Il10, Maf, Ahr, Prdm1, Irf4, Jun, and Ikfz3 were quantified by qPCR (dotted line at 1 indicates the value of control untreated TH17 cells). Data are presented as mean±s.e.m. of five independent experiments.
The hypothesized role of IL-33 in shifting the phenotype of pro-inflammatory TH17 cells toward an anti-inflammatory subset producing IL-10 was further supported by the gene expression profile observed after administration of IL-33 to ex vivo sorted splenic IL-17A-EGFP+RFP− (TH17) cells (Figure 6a). TH17 cells downregulated Rorc upon IL-33 administration, while Foxp3 levels remained unchanged in this cell subset (Figure 6b). On the contrary, Il10 expression was induced by IL-33 in a dose-dependent manner in TH17 cells, while its levels were only marginally, non-significantly increased in Tregs. Furthermore, the expression of Ifng was efficiently downregulated by IL-33 in TH17 cells even at low doses (Figure 6b). Surprisingly, although fresh ex vivo sorted TH17 cells expressed Il17a (control), the transcript could not be detected upon 24 h of culture without any TCR activator, independently of the addition of IL-33 to the culture medium. Finally, we sought to compare the in vitro suppressive capacity of IL-33-stimulated TH17 cells with untreated TH17 cells and professional Tregs. As expected, professional Tregs inhibited T-cell proliferation to the greatest extent. Nevertheless, addition of IL-33 significantly enhanced murine TH17 suppressor functions (Figure 6c and d). Collectively, our data indicate that the exacerbated TH17 response observed in vivo in St2−/− mice upon inflammation, as well as the reduced levels of TH17-derived IL10 observed in anti-ST2-treated mice are due, at least in part, to IL-33 signaling in TH17 cells. This leads to a Foxp3-independent increase in Il10 and a decrease in Rorc and Ifng expression, promoting the acquisition of a regulatory phenotype.

IL-33 upregulates Il10 and downregulates Ifng expression in fresh ex vivo sorted TH17 cells and promotes regulatory functions in TH17 cells in vitro. (a) IL17A-eGFPxFoxp3-mRFP reporter mice (n=7) were injected i.p. with anti-CD3 at days 1, 3, and 5. Splenic TH17 cells (CD45+CD4+IL-17A-GFP+Foxp3-RFP−) and Treg cells (CD45+CD4+IL-17A-eGFP−Foxp3-mRFP+) were sorted 4 h after the last injection. Sorted cells were cultured in the presence of 0, 20, or 100 ng ml−1 of IL-33 for 24 h or directly stored in Trizol at −80 °C (Control). (b) mRNA expression levels of Rorc, Foxp3, Il10, Ifng, and Il17a were measured by qPCR in both subsets. (c) Suppression assay in a mouse T-cell culture was performed by using MACS-enriched CD25+ Treg cells and in vitro generated TH17. TH17 treated or not with 20 ng ml−1 of IL-33 were sorted at day 4 for CD4+IL17eGFP+FoxP3mRFP−. After 3 days of co-culture, frequencies of non-proliferating responder CFSE-labeled CD4+CD25− T cells were quantified by FACS. (d) Bar histogram summarizing frequencies of non-proliferating responder cells obtained in several assays, relative to professional Tregs (100% suppression ability, dotted line). (b) Results are presented as mean±s.e.m. and are representative of three independent experiments. * P<0,05, ** P<0,005, *** P<0,0001 as calculated by one-way ANOVA with Bonferroni post-test. ns, not significant. nd, non-detectable. (c–d) Data are shown as mean±s.e.m. of six independent experiments. **P<0,005 as calculated by paired Student’s t-test.
DISCUSSION
It has recently been demonstrated by us that the SI controls potentially inflammatory TH17 cells to avoid exacerbated immune responses.5 However, the underlying mechanisms governing this process are only partially understood. IL-10 (refs 9,19) and TGF-β (ref. 10) are two key immune-regulatory mediators known to limit TH17 cell pathogenicity, but both cytokines are mostly produced by myeloid cells and are not specifically induced in the SI. Here we identified IL-33 as a local factor produced by epithelial cells in the SI, in control of the TH17 lineage. A proper understanding of the tissue-derived, local factors controlling TH17 cells may help fighting chronic inflammatory processes and autoimmune diseases driven by this T-cell subset, such as multiple sclerosis,2 rheumatoid arthritis,31 and inflammatory bowel disease.3
Using the anti-CD3 model, we describe here for the first time ST2 expression in TH17 cells in vivo and we show that the ST2/IL-33 axis can induce the conversion of TH17 cells from an inflammatory phenotype to a functional suppressive subset co-producing IL-10. In recent years, the IL-33/ST2 axis has emerged as a key modulator of inflammation in epithelial tissues,32 particularly in the gut mucosa. In this study, we show that IECs are the main source of IL-33 upon inflammation. Although IECs are CD3− and therefore cannot be directly activated by CD3-specific antibodies to release IL-33, several studies have shown that anti-CD3 treatment activates CD3+ intestinal intraepithelial lymphocytes, leading to the induction of cytotoxic pathways and consequently to tissue damage.33, 34, 35, 36, 37 IL-33 is released by epithelial cells upon tissue damage and functions as an alarmin, alerting the immune system to mount an appropriate response.14, 38, 39 Results from our group also suggest this indirect mechanism mediated by intraepithelial lymphocytes, by which anti-CD3 administration leads to release of IEC-derived IL-33 (data not shown). Our results show a rapid and strong IL-33 response in the proximal part of the SI, coinciding with the site of local inflammation caused transiently by anti-CD3 treatment. We found that the induction of IL-33 occurred in a gradient-like fashion along the intestine, from proximal to distal, similar to the expression of CCL20 and therefore coinciding with the spatial distribution of TH17 cells in the gut.5 In contrast, IL-33 expression upon treatment in the spleen remains at levels comparable to steady state. We found ST2+ TH17 cells both in the spleen and the SI of anti-CD3-treated animals. However, we detected a lower mean fluorescence intensity of ST2 in intestinal TH17 cells compared with the spleen, indicating a lower expression of the receptor at the per cell basis in the SI. This might indicate a down-modulation of ST2 in TH17 cells upon arrival in the SI, where IL-33 is abundantly expressed. The hypothesized down-modulation, together with the absence of ST2 in TH17 cells at steady state would support the notion of an inducible and transient ST2 expression in this T-cell subset, similar to what has been reported for TH1 cells21 and in contraposition to the constitutive and abundant expression of ST2 shown by TH2 cells and Tregs.40 About 90% of splenic ST2+ TH17 cells co-express CCR6, a receptor known to direct these cells particularly to the proximal part of the SI, the region showing the strongest upregulation of the CCR6-ligand CCL20.5 The differential expression of CCR6 in the intestinal ST2+ (∼50%) and ST2− (∼15%) TH17 population might also indicate that the former represent newly accumulating TH17 cells, as CCR6 is being downregulated in TH17 cells after their recruitment to the gut.5 Our findings indicate a spatial separation of ST2 induction in the spleen and the provision of IL-33 by the gut epithelium, which may be a mechanism to fine-tune the effects of IL-33 on TH17 cells.
It has recently been published that ST2 deficiency led to exacerbated colitis due to deficient iTreg differentiation22 and T cells driving colitis are known to express high amounts of IL-17A and the pro-inflammatory cytokines IFN-γ, TNF-α, and GM-CSF.3, 41, 42, 43 We did find increased Il17a, Ifng, and Tnf levels in the duodenum, as well as decreased levels of the anti-inflammatory mediator Il10, in association with a higher degree of inflammation in the St2−/− mice. Remarkably, ST2 deficiency also led to a specific increase both in frequencies and absolute numbers of TH17 cells (RORγt+ T cells and IL-17A+ T cells) in the SI upon inflammation. In addition, intestinal TH17 cells showed a greater pathogenic phenotype, featured by increased frequencies of both IL-17A+IFN-γ+ and IL-17A+TNF-α+ T cells. We observed equivalent frequencies of Foxp3-expressing cells in St2−/− and WT mice in the SI upon inflammation. However, further analysis on the functionality of Tregs would be needed to exclude any contribution from these regulatory cells on the exacerbated TH17 response found in the absence of ST2. On the one hand, we are aware that the anti-CD3 treatment is well suited to study the plasticity of TH17 responses, but it does not reflect the full complexity of an inflammatory response in the gut, as it occurs during chronic inflammation. This may also explain why the differences observed between St2−/− and WT mice in histopathology are rather mild. On the other hand, IL-10 is an anti-inflammatory cytokine that can be induced in TH17 cells independently of Foxp3 and that confers these cells the intrinsic ability to exert regulatory functions.19 The fact that genetic deletion of St2 and blocking of ST2 in vivo resulted, both, in a decreased LP IL-10+ TH17 population suggests a clear link between ST2 signaling and the secretion of the anti-inflammatory cytokine IL-10 by TH17 cells. Taken together, our study complements the findings of other groups by extending the role of IL-33 beyond the induction and maintenance of Tregs in the colon22 and visceral adipose tissue44 to the conversion of pro-inflammatory into regulatory TH17 cells in the SI.
IL-33 stimulation of ex vivo sorted TH17 cells indeed led to changes in the expression levels of key genes modeling TH17 functionality. Rorc was downregulated, although Foxp3 expression did not change in this subset, indicating that IL-33 limits TH17 cells differentiation ex vivo, similarly to what we observed in vivo in St2−/− mice. We also observed a reduction in Ifng, while Il10 expression was significantly increased, in a dose-dependent manner, upon IL-33 stimulation. Surprisingly, Il17a expression could only be quantified in fresh ex vivo sorted TH17 cells. In line with the ex vivo data, in vitro TH17 cell cultures responded to IL-33 stimulation by downregulating pro-inflammatory genes such as Tbx21, Ifng, and Csf2, as well as upregulating Il10. Induction of IL-10 by the IL-33/ST2 axis in TH17 cells was concurrent with the induction of a combination of transcription factors known to mediate IL-10 production in T cells. Among the transcripts positively regulated upon IL-33 stimulation together with Il10 were Ahr, Ikzf3, Irf4, and Jun. Moreover, Ahr expression correlated with the expression of Il10 in the cultures analyzed and it has already been shown to mediate TH17 cell plasticity, to enhance IL-10 production and to be involved in the conversion of TH17 into regulatory T cells.10 Further experiments will be required to elucidate the role of Ahr in relation to the IL-33/ST2 axis. On a functional level, mouse TH17 cells stimulated with IL-33 were able to suppress the proliferation of responder T cells in vitro, suggesting the acquisition of a regulatory phenotype. Importantly, in human TH17 cells, in vitro IL-33 stimulation promoted an increase in IL-10 production, downregulation of TBX21 expression and reduction of GM-CSF secretion. GM-CSF has been shown to be a key cytokine for T-helper cells involved in chronic immune disorders, as GM-CSF-producing T cells exert a pathogenic role in the inflamed brain of multiple sclerosis patients.45 Although our in vitro data demonstrate a role for the IL-33/ST2 axis in dampening TH17 pathogenicity, we would like to emphasize that we cannot rule out a contribution of other ST2-expressing immune subsets, such as Tregs, Th2, and ILC2 (ref. 46) to the effects that we observe in vivo. Further experiments using inducible ST2 knockout systems will be required to address this question.
In summary, our results extend the spectrum of functions of the alarmin IL-33, highlight once more the role of the gut as an organ with systemic immune-regulatory functions and mechanistically explain the conversion of inflammatory into regulatory TH17 cells occurring in the SI. A better knowledge of these tissue-derived, local factors in control of inflammatory and regulatory T cells might open the door for targeted immune modulation in the treatment of chronic inflammation and autoimmune diseases.
METHODS
Mice. Wild-type C57BL/6 and congenic B6.SJL-Cd45.1 mice were purchased from Charles River. St2−/− mice,23 IL17A-eGFPxFoxp3-mRFP reporter mice,5 and IL-10-GFP reporter mice47 on a C57BL/6 background were bred and housed under specific pathogen-free conditions in the animal facility of the Charité. For some experiments, IL-10-GFP reporter mice were crossed onto St2−/− mice (IL-10-GFP.T1ST2KO). Mice were age- and sex-matched and 8–12 weeks old when used in experiments. Protocols for animal experiments were approved in accordance with institutional, state, and federal guidelines (Landesamt Für Gesundheit und Soziales, Berlin, Germany).
In vivo T-cell stimulation and intestinal lymphocyte isolation. Mice were injected with anti-CD3 antibodies (20 μg, clone 145-2C11, DRFZ) i.p. 1–3 times at an interval of 2 days between injections and were killed 4 h (or 24 h, where indicated) after the final injection. For the controls, isotype control or PBS was injected. Intestinal lymphocytes were isolated as previously described with minor modifications.5 In brief, SIs were removed, opened longitudinally and then were cut into strips 1 cm in length. Tissues were washed twice in PBS and incubated with Hank’s buffered saline (5% FCS) in the presence of 2 mM of EDTA at 37 °C for 30 min on a shaker. Afterwards, tissues were incubated with complete RPMI medium supplemented with 100 U ml−1 of collagenase type IV (Sigma, Steinheim, Germany) at 37 °C for 30–40 min on a shaker. All released cells were loaded onto a Percoll gradient and centrifuged. The cells between 40 and 100% Percoll were collected and used as intestinal lymphocytes.
In vivo blocking of ST2. IL-10-GFP reporter mice (VertX, Jaxmice, Bar Harbor, ME) were injected with anti-ST2 antibodies (50 μg, clone DIH4, Biolegend, Fell, Germany) or isotype control antibodies (rat IgG1, clone RTK2071, Biolegend) i.p. 2 times (according to refs 24,25) 30 min prior to anti-CD3 injection and were killed after the second injection. Intestinal lymphocytes were isolated as described above.
Immunofluorescence microscopy. Small intestines and spleen were removed from wild type or IL17A-EGFPxFoxp3-RFP reporter mice before and after CD3-specific antibody treatment in vivo. Tissues were fixed in 1% paraformaldehyde at 4 °C over night. Subsequently paraformaldehyde was replaced with 30% sucrose solution and incubated at 4 °C for 6–8 h. After that, samples were washed and placed in oct-filled cryomolds. For freezing, the cryomolds were placed in chamber filled with 2-methylbutane, which was subsequently placed in dry ice submerged with ethanol. After freezing, the samples were stored at −80 °C. Cryosections were cut at 7 mm on a Leica CM1850 freezing microtome, transfered onto Superfrost Plus Gold slides (Fisher Scientific, Jena, Germany) and air dried. For immunohistological staining, sections were surrounded with a PAP pen (Zymed Laboratories, Jena, Germany) and submerged with PBS for 20 min. After that, sections were blocked for 20 min at room temperature with 10% rat serum in PBS. For immunofluorescent staining, antibodies were diluted in staining buffer (PBS, 3% BSA, 0.1% Tween, and 0.01%Triton) and incubated at room temperature for 60 min. The following antibodies were used for staining: anti-CD11c-Alexa Fluor 647 (N418, produced in house), anti-PECAM-Alexa Fluor 647 (MEC13.3, Biolegend), anti-IL-33-Alexa Fluor 594 (polyclonal goat IgG, R&D systems, Minneapolis, MN), anti-EpCAM-Alexa-488 (G8.8, Biolegend), Hoechst 33342 or DAPI (as indicated), and anti-GFP-Alexa-488 (polyclonal, Goat IgG, Rockland, Boyertown, PA). Nuclear stains were added after washing once with PBS for 5 min and incubated for 10 min at room temerature. After staining, samples were washed three times by immersing in PBS for 5 min and mounted with ProLong gold mounting medium (Life Technologies, Waltham, MA). Sections were analyzed by confocal microscopy using a Zeiss LSM 710 microscope with × 20/0.5 NA (air) objective at room temperature.
Histopathology. SIs were removed and fixed immediately in 10% neutral-buffered formalin in PBS for >24 h, embedded in paraffin and cut into 5–7 μm sections. The sections were deparaffinized and stained with hematoxylin and eosin to determine histological changes.
Antibodies and intracellular cytokine staining. Anti-CD44-APC, anti-CD62L-FITC, anti-CD4-APC-Cy7, and anti-CD25-PE were purchased from eBioscience and used for FACS sorting of mouse naive T-cell populations. To stain T cells from spleen and SI of IL17A-EGFPxFoxp3-RFP or IL-10-GFP reporter mice before and after CD3-specific antibody treatment, cell suspensions were obtained as described above and first incubated with erythrocyte lysis buffer (ACK), followed by an FC-blocking antibody (anti CD16/32, clone 2.4G2, generated in the DRFZ) to prevent non-specific binding. Single cell suspensions were stained with anti-CD45-Pacific Blue, anti-CD4-BV785, anti-TCRβ-PE-Cy7 anti-CCR6-BV605, live-dead-eF780 (all from eBioscience, Jena, Germany) and anti-ST2 (DJ8, MD Biosciences) conjugated to digoxigenin (produced in the DRFZ). A second incubation with anti-DIG-PE, followed by two rounds of Faser kit-PE (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturers’ protocol was performed. For cytokine detection, cells were restimulated with PMA (5 ng ml−1) and Ionomycin (500 ng ml−1) for 5 h with addition of Brefeldin A and Monensin (5 μg ml−1) at 30 min, followed by fixation in 2% formaldehyde. Intracellular staining was performed in PBS/0.2% BSA containing 0.05% saponin with anti-IL-17A-FITC (TC11-18H10.1, Biolegend) or anti-IL-17A-APC (TC11-18H10.1, Biolegend) anti- IFN-γ-Pacific Blue (XMG1.2, eBioscience) and anti-TNFα-PerCP (MP6-XT22, Biolegend). T-bet, Roryt, c-maf, and Foxp3 protein amounts were analyzed using the Foxp3 staining buffer set (eBioscience) according to the manufacturer’s instructions. Briefly, cells were stained with antibodies against CD4 (RM4-5, PE-Cy7, eBioscience) and CD45.2 (104, APC, BD, Heidelberg, Germany) and then fixed with fixation/permeabilization buffer (BD), followed by intracellular staining with Pacific Blue-conjugated anti-T-bet (4B10, Biolegend), PE-conjugated anti-Rorγt (104 or Q31-378 BD) and FITC-conjugated anti-FoxP3 (FJK-16 s, eBioscience) in permeabilization buffer. The following antibodies were used for FACS sorting of human peripheral blood mononuclear cells: anti-CCR6-PE (11A9) or biotinylated anti-CCR6 followed by streptavidin-Pacific Blue, anti-CCR4-PE (1G1), anti-CXCR3-APC (1C6/CXCR3), anti-CD45RA-FITC (HI100, all from BD Biosciences), anti-CD25-FITC (B1.49.9), anti-CD8-PC5 (B9.11, both from Immunotech, Quebec, Canada).
Flow cytometry and FACS sorting. Mouse peripheral T cells were obtained from spleen and lymph nodes of 8- to 12-week-old mice and CD4+CD62L+CD25−CD44− T cells (naive T cells) were sorted by FACSAria II (BD). Human CCR6+CCR4+CXCR3–CD45RA–CD25–CD8– TH17 cells were sorted according to Zielinski et al. Single cell suspensions were analyzed on a Becton Dickinson Fortessa or LSRII and data analyzed with FlowJo software (Tree Star, Ashland, OR).
TH17 differentiation in vitro. Splenocytes from wild-type C57BL/6 mice were incubated with CD4 microbeads and then positively selected through LS columns (Miltenyi Biotec). After enrichment, naive cells (CD4+CD25−CD62LhiCD44low) were FACS sorted as mentioned above. CD4+ naive T cells were cultured for 5 days at 106 cells ml−1 with plate bound anti-CD3 (3 μg ml−1; 145-2C11; Miltenyi) and soluble anti-CD28 (2 μg ml−1; 37.51; Miltenyi) in RPMI Media 1640 (Invitrogen, Jena, Germany) supplemented with 10% fetal calf serum (Hyclone, Logan, UT), 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 5 mM 2-β-mercaptoethanol under TH17 conditions: TGF-β (2 ng ml−1; R&D systems) and IL-6 (25 ng ml−1; R&D systems). Neutralizing anti-IFN-γ (XMG1.2; own produced) and anti-IL-4 (11B11; produced in house) were each used at 5 μg ml−1. Where indicated, IL-33 (20 ng ml−1; R&D) was added to the culture.
For the human cultures, healthy leukapheresis blood and buffy coats were randomly obtained from the Charité Blood Bank and Deutsches Rotes Kreuz (DRK). Ethics approval was obtained from the Institutional Review Board of the Charité-Universitätsmedizin Berlin, Germany (EA1/221/11, EA1/293/12). All work was carried out in accordance with the Declaration of Helsinki for experiments involving humans. Human peripheral blood mononuclear cells were isolated using Ficoll-Paque Plus (GE Healthcare, Salt Lake City, UT) and T cells were isolated with CD4 microbeads (Miltenyi Biotech), followed by the above-mentioned sorting strategy.
Suppression assay. CFSE (5 μg ml−1) labeled CD4+CD25− T cells (responder cells) were cultured in 96-well round bottom plates at 2 × 104 or 1 × 104cells per well with 8 × 105 irradiated APCs (splenocytes MACS depleted for CD4+, CD8+, and CD25+ T cells) as feeder cells in the presence of 2 × 104 cells per well of FACS-sorted CD4+IL-17A−Foxp3+ (Tregs) or CD4+IL-17A+Foxp3− T cells (TH17), previously cultured for 4 days in the presence or absence of IL-33 (20 ng ml−1). Cell cultures were stimulated with 0.1 μg ml−1 of anti-CD3 antibody (2C11, Miltenyi). After 4 days, cells were collected, stained, and the CFSE signal was analyzed by flow cytometry.
RT-PCR. RNA was extracted according to the manufacturers’ protocol (Rneasy, Qiagen, Hilden, Germany) and complementary DNA was synthesized using Taqman RT reagents (Applied Biosystems, Waltham, MA), following the manufacturer’s protocol. Quantitative real-time PCR was performed in triplicate using TaqMan Gene Expression Assays (Applied Biosystems) for the respective genes on StepOnePlus Real-Time PCR System (Applied Biosystems). mRNA levels were normalized to HPRT as a housekeeping gene for mouse samples and to GAPDH for human samples and differences were calculated using the 2ΔCt method.
Gene expression analysis. Total RNA extracted (100 ng; RNeasy, Qiagen) from intestinal CD45neg cells from CD3-specific antibody-treated or untreated animals were used to perform a genome-wide transcriptional profiling assay (GeneChip Mouse 2.0 ST Array, Affymetrix, Santa Clara, CA). Data was analyzed with GeneSpring GX 11 (Agilent Technologies, Santa Clara, CA).
Statistical analysis. Where appropriate, Student’s t-test was used. For the comparison of more than two groups a one-way ANOVA followed by Bonferroni multiple comparison test was performed. All statistical analysis was calculated in Prism (GraphPad). Differences were considered to be statistically significant when P<0.05.
References
Littman, D.R. & Rudensky, A.Y. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140, 845–858 (2010).
Kebir, H. et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175 (2007).
Elson, C.O. et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 (2007).
Basu, R., Hatton, R.D. & Weaver, C.T. The Th17 family: flexibility follows function. Immunol. Rev. 252, 89–103 (2013).
Esplugues, E. et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011).
Chatenoud, L. & Bluestone, J.A. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat. Rev. Immunol. 7, 622–632 (2007).
Musch, M.W. et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J. Clin. Invest. 110, 1739–1747 (2002).
Chatenoud, L., Bach, J.-F.O. & Sakaguchi, S. CD3-specific antibodies restore self-tolerance: mechanisms and clinical applications This review comes from a themed issue on autoimmunity edited. Curr. Opin. Immunol. 17, 632–637 (2005).
Huber, S. et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565 (2011).
Gagliani, N. et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225 (2015).
Chewning, J.H. & Weaver, C.T. Development and survival of Th17 cells within the intestines: the influence of microbiome- and diet-derived signals. J. Immunol. 193, 4769–4777 (2014).
Sefik, E. et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997 (2015).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005).
Moussion, C., Ortega, N. & Girard, J.-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE 3, e3331 (2008).
Haraldsen, G., Balogh, J., Pollheimer, J., Sponheim, J. & Küchler, A.M. Interleukin-33 – cytokine of dual function or novel alarmin? Trends Immunol. 30, 227–233 (2009).
Kakkar, R. & Lee, R.T. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 7, 827–840 (2008).
Molofsky, A.B., Savage, A.K. & Locksley, R.M. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 42, 1005–1019 (2015).
Kole, A. & Maloy, K.J. Interleukin-10 in health and disease. Curr. Topics Microbiol. Immunol. 380, 19–38 (2014).
Evans, H.G. et al. TNF-alpha blockade induces IL-10 expression in human CD4+ T cells. Nat. Commun. 5, 3199 (2014).
Löhning, M. et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl Acad. Sci. USA 95, 6930–6935 (1998).
Baumann, C. et al. T-bet– and STAT4–dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc. Natl Acad. Sci. USA 112, 4056–4061 (2015).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Townsend, M.J., Fallon, P.G., Matthews, D.J., Jolin, H.E. & McKenzie, A.N. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J. Exp. Med. 191, 1069–1076 (2000).
Hashiguchi, M. et al. IL-33 activates eosinophils of visceral adipose tissue both directly and via innate lymphoid cells. Eur. J. Immunol. 45, 876–885 (2015).
Sedhom, M.a.K. et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62, 1714–1723 (2013).
Murakami-Satsutani, N. et al. IL-33 promotes the induction and maintenance of Th2 immune responses by enhancing the function of OX40 ligand. Allergol. Int. 63, 443–455 (2014).
Zielinski, C.E. et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484, 514–518 (2012).
Stumhofer, J.S. et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 8, 1363–1371 (2007).
Li, P. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012).
Xu, J. et al. c-Maf regulates IL-10 expression during Th17 polarization. J. Immunol. 182, 6226–6236 (2009).
Hirota, K. et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 204, 41–47 (2007).
García-Miguel, M., González, M.J., Quera, R. & Hermoso, M.A. Innate immunity modulation by the IL-33/ST2 system in intestinal mucosa 2013, 1–13 (2013).
Swamy, M. et al. Intestinal intraepithelial lymphocyte activation promotes innate antiviral resistance. Nat. Commun. 6, 7090 (2015).
Merger, M. et al. Defining the roles of perforin, Fas/FasL, and tumour necrosis factor alpha in T cell induced mucosal damage in the mouse intestine. Gut 51, 155–163 (2002).
Ogata, M. et al. DNA repair after DNA fragmentation in mouse small intestinal epithelial cells. Cell Tissue Res. 335, 371–382 (2009).
Yaguchi, K. et al. DNA fragmentation and detachment of enterocytes induced by anti-CD3 mAb-activated intraepithelial lymphocytes. Cell Tissue Res. 315, 71–84 (2004).
Ogata, M., Ota, Y., Nanno, M., Suzuki, R. & Itoh, T. Activation of intra-epithelial lymphocytes; their morphology, marker expression and ultimate fate. Cell Tissue Res. 356, 217–230 (2014).
Bonilla, W.V. et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science 335, 984–989 (2012).
Oboki, K. et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl Acad. Sci. USA 107, 18581–18586 (2010).
Peine, M., Marek, R.M. & Löhning, M. IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol. 37, 321–333 (2016).
Stallhofer, J. et al. Lipocalin-2 is a disease activity marker in inflammatory bowel disease regulated by IL-17A, IL-22, and TNF-α and modulated by IL23R genotype status. Inflamm. Bowel Dis. 21, 2327–2340 (2015).
Sabatino, A., Di Lenti, M.V., Giuffrida, P., Vanoli, A. & Corazza, G.R. New insights into immune mechanisms underlying autoimmune diseases of the gastrointestinal tract. Autoimmun. Rev. 14, 1161–1169 (2015).
Griseri, T. et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity 43, 187–199 (2015).
Vasanthakumar, A. et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16, 276–285 (2015).
Noster, R. et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 6, 241ra80–241ra80 (2014).
Neill, D.R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Madan, R. et al. Non-redundant roles for B cell-derived IL-10 in immune counter- regulation. J. Immunol. 183, 2312–2320 (2009).
Acknowledgements
The authors thank Professor McKenzie (MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus) for providing the St2-deficient mice. We are grateful to T. Kaiser and J. Kirsch for essential flow cytometry support and the staff of the DRFZ and CCO animal facilities for animal care. We are also grateful to the DRFZ Lab managers for their assistance with antibodies and reagents. We also want to thank N. Gagliani for assistance and support. Funding: A.E.H. is supported by grants from the DFG (EXC 257 NeuroCure, HA5354/6-1 and HA5354/8-1). E.E. is supported by grants from the Leona M. and Harry B. Helmsley Charitable Trust, the Crohn’s & Colitis Foundation of America (#311143) and from the DFG (EXC 257 NeuroCure and SFB633).
Author contributions
A.P.-R., A.E.H., J.B.S. and E.E. designed the study, A.P.-R., J.B.S., C.B., D.C.-S., C.C., R.N., P.P, S.H. and C.E.Z. performed experiments, M.L. provided reagents. A.P.-R., A.E.H. and E.E. wrote the manuscript. All authors analyzed data and interpreted results.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Pascual-Reguant, A., Bayat Sarmadi, J., Baumann, C. et al. TH17 cells express ST2 and are controlled by the alarmin IL-33 in the small intestine. Mucosal Immunol 10, 1431–1442 (2017). https://doi.org/10.1038/mi.2017.5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.5
This article is cited by
-
The role of T cells in age-related diseases
Nature Reviews Immunology (2022)
-
The interleukin-33 receptor contributes to pulmonary responses to ozone in male mice: role of the microbiome
Respiratory Research (2019)
-
The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer
Mucosal Immunology (2019)
