
Abstract
Interferon regulatory factor 5 (IRF5) is a key transcription factor involved in the control of the expression of proinflammatory cytokine and responses to infection, but its role in regulating pulmonary immune responses to allergen is unknown. We used genetic ablation, adenoviral vector-driven overexpression, and adoptive transfer approaches to interrogate the role of IRF5 in pulmonary immunity and during challenge with the aeroallergen, house dust mite. Global IRF5 deficiency resulted in impaired lung function and extracellular matrix (ECM) deposition. IRF5 was also essential for effective responses to inhaled allergen, controlling airway hyperresponsiveness, mucus secretion, and eosinophilic inflammation. Adoptive transfer of IRF5-deficient alveolar macrophages into the wild-type pulmonary milieu was sufficient to drive airway hyperreactivity, at baseline or following antigen challenge. These data identify IRF5-expressing macrophages as a key component of the immune defense of the airways. Manipulation of IRF5 activity in the lung could therefore be a viable strategy for the redirection of pulmonary immune responses and, thus, the treatment of lung disorders.
Similar content being viewed by others


Introduction
Asthma is a heterogeneous disease of the airways characterized by airway remodeling, mucus production, airway hyperresponsiveness (AHR), and inflammation. Although asthma has traditionally been considered to be a T helper cell type 2 (Th2)-driven disease, many T cell-directed therapies have not been effective in clinical trials.1, 2 The lack of effective therapies for some types of asthma might reflect the poor understanding of the specific immunological pathways that lead to the disease. Interferon regulatory factor 5 (IRF5) is a key transcription factor involved in the control of the expression of proinflammatory cytokine responses to microbial infection and type I interferon responses to viral infection.3 IRF5 mRNA expression is increased in response to interferon-γ (IFN-γ) and granulocyte-macrophage colony-stimulating factor,4 whereas activation of IRF5 protein occurs in response to immune signaling pathways, such as downstream of TLRs and so on, via posttranslational modifications such as phosphorylation and ubiqutination.5 Polymorphisms in the gene encoding IRF5 that lead to higher mRNA expression are associated with many autoimmune diseases, such as rheumatoid arthritis,6 Sjogrens syndrome,7 systemic lupus erythematosus,8 multiple sclerosis,7 and inflammatory bowel disease.9 Emerging genetic studies have provided evidence that implicates IRF5 in the pathogenesis of allergic disease. Wang et al.10 identified a common IRF5 haplotype to be associated with asthma and the severity of asthmatic symptoms; furthermore, the risk associated with IRF5 was found to be opposite in direction to those for autoimmune disorders. Recently, we demonstrated that IRF5 is critical in establishing inflammatory macrophage phenotypes involved in the positive regulation of Th1/Th17-associated mediators, such as interleukin (IL)-1, IL12β, IL-23, and tumor necrosis factor-α and the negative regulation of Th2-associated mediators, such as IL-10.3, 4
Macrophages are the most abundant immune-cell type present in the lung environment under homeostatic conditions and are therefore strategically positioned to control the innate defense of the airways.11, 12 Two defined populations of macrophages in the lung are characterized by unique properties and functions.13 Alveolar macrophages (AMs) exist in the airway lumen and are characterized by high expression of CD11c but lack CD11b expression.14 Interstitial macrophages reside in the lung parenchyma, express high levels of CD11b, low levels of CD11c, and have been shown to control endotoxin-induced airway inflammation.15 As a critical component of pulmonary immunity, AMs are tightly regulated in order to preserve homeostasis; however, the molecular mechanisms by which this occurs are not well understood.
We find that macrophages are the predominant IRF5-expressing cell type in the pulmonary compartment under homeostatic conditions. The absence of IRF5 during exposure to the clinically relevant allergen, house dust mite (HDM), resulted in increased AHR and allergic inflammation, concomitant with elevated expression of extracellular matrix (ECM) molecules and collagen deposition. Significantly, adoptive transfer of IRF5-deficient AMs into the wild-type (WT) milieu was sufficient to drive airway hyperreactivity, even in the absence of antigen stimulation. Conversely, overexpression of IRF5 resulted in ablated AHR, diminished eosinophilia, and decreased type 2 cytokine production. These data identify IRF5-expressing macrophages as a key component of the immune defense of the airways. Manipulation of IRF5 activity in the lung could thereby be a viable strategy for the redirection of pulmonary immune responses and thus the treatment of lung disorders.
Results
IRF5 deficiency leads to augmented type 2 responses in the lung after HDM exposure
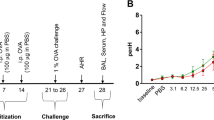
In order to determine the role of IRF5 in regulating the pulmonary environment, we first exposed mice to the aeroallergen, HDM, instilled directly into the airways (Figure 1a). Ablation of IRF5 had a profound effect on airway function, as IRF5−/− mice were hyperresponsive to inhaled methacholine challenge, showing increased airway resistance following 3 weeks of HDM exposure (Figure 1b,c). Phosphate-buffered saline (PBS)-treated IRF5−/− mice displayed increased airway resistance in comparison with WT controls even in the absence of allergen stimulation (Figure 1b,c). Examination of cellular inflammation revealed that total cellular infiltrate and eosinophila were enhanced in the bronchoalveolar lavage (BAL; Figure 1d), but not lung (Figure 1e), of IRF5-deficient mice; analysis of hematoxylin/eosin-stained lung sections confirmed these observations (Figure 1f). Furthermore, IL-13 secretion into the airspaces (Figure 1g) and lung eotaxin-2 levels (Figure 1h) were enhanced in IRF5-deficient animals after week 3 of allergen exposure, as were the expression levels of IL-4, -5, -13, and eotaxin-2 (Supplementary Figure S1A online). Moreover, AMs in the BAL of PBS-treated IRF5−/− mice displayed higher intracellular level of IL-13 in comparison with WT controls (Supplementary Figure S1B) even in the absence of IL-13 secretion in the BAL (Figure 1g), implying a direct effect of IRF5 on inflammatory response in AMs. In addition, there was a significant increase in the population of IL-13+ CD4+ T lymphocytes (Figure 1i). However, there was no difference in total Th1, Th17, and total innate lymphoid (ILC) populations (Supplementary Figure S1C), or in IL-13+-producing ILC2 cells (Supplementary Figure S1D). Interestingly, we detected a small number of IL-17+-producing ILC3 cells in the lung of WT animals and this population was reduced to baseline levels in the IRF5−/− mice (Supplementary Figure S1D). Although ILC3 populations have been shown to be important for obesity-associated deteriorations in lung function,16 their absence in this model did not result in a reduction in AHR. We also observed augmented levels of HDM-specific IgE (Supplementary Figure S1E), no change in HDM-IgG1 (Supplementary Figure S1F) responses, and decreased levels of HDM-IgG2a in IRF5−/− mice (Supplementary Figure S1G); these humoral responses are consistent with previously reported findings.17, 18 Taken together, all classical indicators of allergic immune responses were exacerbated in HDM-exposed IRF5-deficient mice concomitant with airway inflammation and AHR.

Interferon regulatory factor 5 (IRF5) is a critical component of pulmonary homeostasis and type 2 responses after house dust mite (HDM) exposure. (a) Experimental design of HDM-induced allergic airway disease. Resistance (RI) measured in tracheotomized animals to a dose response (b) or at a representative dose of 30 mg ml−1 of methacholine (c). Differential cell counts of Wright-Giemsa-stained cytospins recovered from the (d) bronchoalveolar lavage (BAL) and (e) lung. (f) Lung sections stained with hematoxylin/eosin (H&E); original magnification × 40; scale bar=50 μm, representative photomicrographs are shown. Interleukin-13 (IL-13) levels in the (g) BAL and (h) lung eotaxin-2 and as determined by enzyme-linked immunosorbent assay (ELISA). (i) CD4+T1/ST2+IL-13+ T helper cell type 2 (Th2) cells recovered from the lung and quantified by flow cytometry. Data shown represent mean±s.e.m., *P<0.05, **P<0.01, ****P<0.0001 wild-type (WT) compared with IRF5−/− animals by Mann–Whitney test. Data were generated from four independent experiments; n=7–20 per group.
Pulmonary IRF5 controls airway remodeling
As type 2 responses were enhanced in IRF5-deficient mice in comparison with WT controls (Figure 1), we next examined whether these changes resulted in alterations in mucus secretion and airway remodeling. Goblet cell hyperplasia was increased in HDM-treated IRF5−/− mice in comparison with WT controls (Figure 2a, c). Muc5b, but not Muc5ac, was significantly elevated after allergen exposure (Supplementary Figure S2A,B). Interestingly, a recent study by Roy et al.19 demonstrated that murine Muc5b, but not Muc5ac, is required for mucociliary clearance and for maintaining homeostasis in the murine lung. Moreover, IRF5−/− mice exhibited increased ECM deposition around the airways, in comparison with WT controls, as indicated by Sirius Red staining (Figure 2b,d,e). These alterations were also accompanied by an enhanced M2 (Supplementary Figure S2C,D) and a reduced M1 (Supplementary Figure S2E) signature in the lung. These data indicate that IRF5 deficiency leads to enhanced ECM deposition and thus normal airway function and homeostasis is disrupted. Therefore, our data demonstrate a novel role for IRF5 in regulating airway function via enhanced type 2 skewing (Figure 1), in addition to mucus and collagen production in response to HDM (Figure 2).

Interferon regulatory factor 5 (IRF5) deficiency leads to augmented mucus and collagen production in the lung after house dust mite (HDM) exposure. Lung sections stained with (a) periodic acid–Schiff (PAS) and (b) Sirius Red, respectively. PAS staining to identify mucin-containing cells (purple, top panel); Sirius Red staining of lung sections depicts peribronchiolar and perivascular collagen (red, bottom panel), original magnification × 40; scale bar=50 μm, representative photomicrographs are shown. (c) Scoring of lung sections stained with PAS. (d) Recently synthesized total lung collagen was quantified by a biochemical (Sircol) assay. (e) Quantitative image analysis of subepithelial peribronchiolar collagen density determined by measuring Sirius Red-stained collagen in lung sections under polarized light; data were normalized to wild-type (WT) untreated control and expressed as percent (%) increase. Data shown represent mean±s.e.m., *P<0.05, **P<0.01, WT compared with IRF5−/− animals by Mann–Whitney test. Box–and-whisker plots represent the mean, interquartile range (IQR), and minimum and maximum values. Data were generated from four independent experiments; n=15–20 per group.
IRF5 deficiency impairs pulmonary homeostasis via increased ECM deposition and IL-13 production
We have previously demonstrated that IRF5 expression is a key determinant of inflammatory macrophage phenotype;4 macrophages are the predominant IRF5-expressing cell type in the pulmonary compartment under homeostatic conditions (Figure 3a). IRF5 appeared to be expressed in alveolar epithelium (Supplementary Figure S3B); however, by analyzing cells obtained from BAL we found that the IRF5-expressing cells in the airway lumen, the optimum position to influence airway function, were almost exclusively AMs (Figure 3b and Supplementary Figure S3A,B). As IRF5-expressing AMs were the predominant IRF5-expressing cells present in the airway lumen, and the role of pulmonary macrophage subtypes in maintenance of airway function is unknown, we next investigated whether adoptive transfer of IRF5−/− AMs into the WT airway could reproduce the phenotype we observed in IRF5−/− mice. Adoptive transfer of WT or IRF5−/− macrophages directly to the lungs of WT mice (Figure 3c) did not result in a pulmonary inflammatory infiltrate within the timescale tested (Figure 3d,e); donor cells could be recovered from the airway lumen 24 h after transfer (Supplementary Figure S3C). However, transfer of IRF5−/− AMs into a WT lung environment resulted in a significant alteration in normal airway resistance (Figure 3f). These changes were accompanied by an increase in levels of IL-13 in the airway lumen as determined by enzyme-linked immunosorbent assay (Figure 3g) and the expression of collagen-1α1 (Figure 3h) and fibronectin (Figure 3i) in whole lung tissue homogenates. In order to further investigate whether the observed changes in airway resistance were due to macrophage-driven IL-13 production, we prophylactically treated WT mice with either an IL-13-blocking antibody or isotype control before adoptive transfer of IRF5−/− AMs (Figure 3j). Consistent with our previous observations, blockade of IL-13 was sufficient to ameliorate IRF5−/− macrophage-driven lung function changes, in comparison with isotype control-treated mice (Figure 3k). Thus, delivery of IRF5-deficient AMs is sufficient to influence the pulmonary environment and recapitulate, in a WT lung, the pathophysiological changes observed in IRF5−/− mice in an IL-13-dependent manner.

Interferon regulatory factor 5 (IRF5)-deficient macrophages (MΦ) are profibrotic. (a) Flow cytometric analysis of lung F4/80-positive cells recovered from whole lung homogenates, without bronchoalveolar lavage (BAL). (b) Flow cytometric analysis of lung F4/80-positive cells recovered from BAL. (c) Schematic for adoptive transfer model. Total counts of cells recovered from the (d) BAL and (e) lung. (f) Resistance (RI) measured in tracheotomized animals at a 300 mg ml−1 dose of methacholine. (g) BAL interleukin-13 (IL-13) levels as determined by enzyme-linked immunosorbent assay (ELISA). Expression of (h) collagen-1α1 and (i) fibronectin after methacholine challenge in whole lung homogenates as measured by quantitative PCR (qPCR), and fold changes are calculated relative to naive wild-type (WT) controls. (j) Dosing regimen and experimental design for IL-13-blocking experiment. (k) Resistance (RI) measured in tracheotomized animals at a 300 mg ml−1 dose of methacholine. Data shown represent mean±s.e.m. *P<0.05, WT compared with IRF5−/− animals by Mann–Whitney test. Box-and-whisker plots represent the mean, interquartile range (IQR), and minimum and maximum values. Data were generated from two independent experiments; n=4–10 per group. AM, alveolar macrophage; DC, dendritic cell; IM, interstitial macrophage; IN, intranasal; IP, intraperitoneal.
IRF5-expressing AMs control responses to inhaled aeroallegen
As the key features of allergic airways disease were exacerbated in HDM-exposed IRF5-deficient mice in comparison with controls (Figures 1 and 2) and IRF5-expressing AMs were capable of controlling lung homeostasis (Figure 3), we next examined whether IRF5-expressing AMs were involved in responses to inhaled aeroallergen. To achieve this, we adoptively transferred WT or IRF5−/− macrophages directly to the lungs of WT mice followed by HDM administration (Figure 4a). HDM-exposed IRF5−/−-AM recipients had heightened airway hyperreactivity to methacholine (Figure 4b), as well as increased cellular inflammation in the BAL (Figure 4c) and lung (Figure 4d), increased BAL eosinophilia (Figure 4e), increased lung IL-4 levels (Supplementary Figure S4A), and augmented IL-13 levels in both the BAL (Figure 4f) and lung (Figure 4g) in comparison with HDM-exposed WT-AM recipients. These data indicate that IRF5-expressing AMs regulate responses to inhaled aeroallergen via Th2 cytokines, in particular IL-13.

Interferon regulatory factor 5 (IRF5)-expressing alveolar macrophage (AMs) control responses to inhaled aeroallegen. (a) Schematic for adoptive transfer model. MΦ, macrophage. (b) Resistance (RI) measured in tracheotomized animals at a 300 mg ml−1 dose of methacholine. Total counts of cells recovered from the (c) bronchoalveolar lavage (BAL) and (d) lung. Total numbers of eosinophils recovered from the (e) BAL and (f) interleukin-13 (IL-13)-producing innate lymphoid cells recovered from the lung. (g) BAL and (h) lung IL-13 levels as determined by enzyme-linked immunosorbent assay (ELISA). Data shown represent mean±s.e.m. *P<0.05, wild-type (WT) compared with IRF5−/− animals by Mann–Whitney test. Box-and-whisker plots represent the mean, interquartile range (IQR), and minimum and maximum values; n=4–6 per group. **P<0.01.
Overexpression of IRF5 ameliorates HDM-mediated AHR
Having shown that IRF5 deficiency resulted in dramatic effects on pulmonary immunity both basally and during allergen exposure, we next sought to determine the effect of overexpression of IRF5 in the airway lumen. First, we determined an appropriate intranasal dose of an adenoviral vector expressing IRF5 (AdIRF5) or empty vector (AdC) that would induce augmented IRF5 expression in the lungs (Supplementary Figure S5A,B) without producing an associated inflammatory response (Supplementary Figure S5C–F). Moreover, we observed no changes in cellular influx in the BAL of mice administered with the IRF5 encoding vector compared with the control vector (Supplementary Figure S5G). Next, we used this optimized dosing regimen to generate mice overexpressing pulmonary IRF5 and then exposed them to HDM extract intranasally (Figure 5a). At 2 days after instillation with AdIRF5, AMs expressed human IRF5, whereas Interstitial macrophages did not appear to take up the virus at this time point (Supplementary Figure S5H), consistent with previously observed rapid internalization of adenovirus by AMs.20 After 3 weeks of allergen exposure, we observed amelioration of airway hyperreactivity in mice exposed to AdIRF5 in comparison with mice treated with AdC (Figure 5b,c). Cell infiltration in the airway lumen was unaffected by IRF5 overexpression (Figure 4d). The observed changes in lung function in AdIRF5 mice were accompanied by reduced recruitment of eosinophils to the airway tissue (Figure 5e). Interestingly, in the IRF5−/− model we see these cellular changes in the BAL rather than the lung tissue (Figure 1d,e). This discrepancy in cellular trafficking is likely because of differences in models or systems (transient overexpression 2 days before challenge vs. global deficiency). Moreover, hematoxylin/eosin staining revealed reduced inflammation around the airways in AdIRF5-treated mice (Figure 5f,g) in addition to diminished levels of type 2 chemokines and cytokines such as IL-13 (Figure 5h) and eotaxin-2 (Figure 5i).

Overexpression of interferon regulatory factor 5 (IRF5) ameliorates house dust mite (HDM)-mediated airway hyperresponsiveness (AHR) and inflammation. (a) Experimental design of HDM-induced allergic airways disease. Resistance measured in tracheotomized animals to a dose response (b) or at a representative dose of 30 mg ml−1 of methacholine (c); n=4–5 per group. Differential cell counts of Wright-Giemsa-stained cytospins recovered from the (d) bronchoalveolar lavage (BAL) and (e) lung; n=4–5 per group. (f) Lung sections stained with hematoxylin/eosin (H&E); original magnification × 40; scale bar=50 μm, representative photomicrographs are shown. (g) Semiquantitative scoring of (i) H&E sections. Lung (h) interleukin-13 (IL-13) and (i) eotaxin-2 levels as determined by enzyme-linked immunosorbent assay (ELISA). Data shown represent mean±s.e.m., *P<0.05, **P<0.01, wild-type (WT) compared with IRF5−/− animals by Mann–Whitney test. Box-and-whisker plots represent the mean, interquartile range (IQR), and minimum and maximum values. Data were generated from three independent experiments; n=10–15 per group.
Overexpression of IRF5 results in ameliorated airway remodeling after HDM treatment
Goblet cell hyperplasia was significantly diminished in IRF5-overexpressing mice, as determined by periodic acid–Schiff staining and mucus scoring (Figure 6a,c); however, airway remodeling was not affected by IRF5 overexpression at the time point investigated (Figure 6b,d). These data indicated that overexpression of IRF5 results in an enhanced ability to regulate immune responses in the lung following allergen exposure, as augmented expression of IRF5 resulted in ablated AHR, diminished the production of type 2 cytokines, and decreased eosinophilia after HDM challenge. A complex network of immune pathways regulate the lung environment and disruption of this system by inhaled particles leads to a break in homeostasis; our data indicate that IRF5-expressing macrophages play a central role in the regulation of these responses.

Overexpression of interferon regulatory factor 5 (IRF5) deficiency leads to reduced mucus production in the lung after house dust mite (HDM) exposure. Lung sections stained with (a) periodic acid–Schiff (PAS) and (b) Sirius Red, respectively. PAS staining to identify mucin-containing cells (purple, top panel); Sirius Red staining of lung sections depicts peribronchiolar and perivascular collagen (red, bottom panel), original magnification × 40; scale bar=50 μm, representative photomicrographs are shown. (c) Scoring of lung sections stained with PAS. (d) Quantitative image analysis of subepithelial peribronchiolar collagen density determined by measuring Sirius Red-stained collagen in lung sections under polarized light; data were normalized to AdC phosphate-buffered saline (PBS) control and expressed as percent (%) increase. Data shown represent means±s.e.m. from two individual experiments *P<0.05, wild-type (WT) compared with IRF5−/− animals by Mann–Whitney test. Box-and-whisker plots represent the mean, interquartile range (IQR), and minimum and maximum values. Data were generated from three independent experiments; n=10–15 per group.
Discussion
A key function of the pulmonary tract is the ability to maintain immune homeostasis despite continuous exposure to inhaled antigens. Although macrophages are ideally placed to contribute to the fine-tuning of the pulmonary immune system, the mechanism by which this is achieved is thus far unknown. This work defines a previously undescribed immune pathway by which IRF5-expressing macrophages can directly regulate the lung environment. IRF5 expression in AMs controls pulmonary function as deletion of IRF5 leads to the establishment of a fibrotic phenotype, without affecting levels of transforming growth factor-β (TGF-β) or IL-10 expression in the lung (Figure 2 and Supplementary Figure S5). Adoptive transfer of IRF5-deficient AMs into a WT environment is sufficient to induce AHR and airway remodeling, leading to IL-13 secretion under steady-state conditions (Figure 3). Of interest, several laboratories have described IL-13-producing macrophages in the context of chronic obstructive pulmonary disease,4, 21 fibrotic disease,22, 23, 24 and respiratory syncytial virus infection19, 21, 25 after particle inhalation26 or in response to IL-25 or IL-33.27 Interestingly, a recent report by Saigusa et al.28 described a role for IRF5 as a positive regulator of key genes involved in fibrosis in dermal fibroblasts at steady state and after bleomycin challenge.26 Dalmas et al.29 found IRF5 to be negatively associated with collagen deposition and remodeling in adipose tissue.27 Together, these studies indicate that IRF5 plays a role in driving fibrotic processes, but these roles may be pleiotropic depending on the tissue and context.
IRF5 is a key component of IFN signaling and there is evidence to suggest that the type 1 IFN system is involved in pathogenesis of Th2 disease, as reduced IFN signatures have been observed in asthma patients in comparison with controls.26, 27, 30 Interestingly, our work shows that IRF5 appears to control aspects of allergic airways disease, as IRF5 overexpression appears to shift the inflammatory profile in the lung from eosinophilic inflammation toward a neutrophilic signature. Recently, we demonstrated that IRF5 is critical in establishing inflammatory macrophage phenotypes involved in the positive regulation of Th1/Th17-associated mediators and the negative regulation of Th2-associated mediators such as IL-10.3, 4 Interestingly, in models of lupus and obesity, respectively, IRF5-deficient mice were associated with increased expression of type 2 cytokines.18 Recently, Wang et al.10 identified a common IRF5 haplotype to be associated with asthma and the severity of asthmatic symptoms and furthermore, the risk associated with IRF5 was found to be opposite in direction to those for autoimmune disorders; our data provide mechanistic evidence to support these findings. Our previous data have demonstrated a critical role for IRF5 in defining the M1 lineage, based on largely in vitro systems. In vivo, macrophages are highly plastic and, during pulmonary inflammatory disease macrophage populations are capable of finely tuning their activity and develop mixed phenotypes.30, 31, 32 Our data here indicate that although pulmonary macrophages express M2 markers, they also express IRF5 and are thus primed to adapt to various environmental signals.
In summary, this work defines a novel pathway by which alterations in IRF5 expression resulted in the development of AHR. We outline a vital role for IRF5 in mediating pulmonary immunity and dictating allergic immune responses in the lung. The novel relationships between IRF5, lung immunity, and AHR reported here have the potential to affect therapies for asthma and highlight IRF5 as a molecular target for the treatment of pulmonary disease.
Methods
Animals. Female WT or IRF5−/− mice on a C57BL/6 background, 6–8 weeks old, received 25 μg HDM extract (Dermatophagoides pteronyssinus in PBS, Greer Laboratories, Lenoir, NC) or 25 μl PBS intranasally 5 days a week for the indicated durations. UK Home Office guidelines for animal welfare based on the Animals (scientific procedures) act 1986 were strictly observed. The Imperial College London Animal Welfare and Ethical Review Body approved this protocol. All surgeries were performed under ketamine and sodium pentobarbital anesthesia and all efforts were made to minimize suffering.
Measurement of AHR. Airway responsiveness was determined by direct measurements of resistance and compliance in anesthetized and tracheotomized mice in response to inhaled PBS or methacholine (Sigma, Cambridge, UK), at the indicated doses, for 1 min in an EMMS system (EMMS, Hampshire, UK).
Cell preparations. In order to obtain BAL, the airways of the mice were lavaged three times with 0.4 ml of PBS via a tracheal cannula. BAL cell counts are quoted number per ml of recovered BAL fluid. BAL fluid was centrifuged (700 g, 5 min, 4 °C); cells were resuspended in 0.5 ml complete media (RPMI+10% fetal calf serum, 2 mM L-glutamine, 100 U ml−1 penicillin/streptomycin). Cells were counted and pelleted onto glass slides by cytocentrifugation (5 × 104 cells per slide). Differential cell counts were performed on Wright-Giemsa-stained cytospins. Percentages of eosinophils, lymphocyte/mononuclear cells, neutrophils, and macrophages were determined from a total of 400 cells. To obtain absolute numbers of each leukocyte subtype, these percentages were multiplied by the total number of cells obtained in the lavage fluid.
To disaggregate the cells from the lung tissue, one finely chopped left lobe of lung was incubated at 37 °C for 1 h in digest reagent (0.15 mg ml−1 collagenase type D, 25 μg ml−1 DNase type I) in complete RPMI media. The recovered cells were filtered through a 70-μm nylon sieve, washed twice, resuspended in 1 ml complete media, and counted in a hemocytometer before cytocentrifugation; lung cell counts are quoted as total cell number per ml of this suspension. Differential cell counts were performed on Wright-Giemsa-stained cytospins. To obtain absolute numbers of each leukocyte subtype, these percentages were multiplied by the total number of cells recovered. All differential counts were performed blind and in a randomized order at the end of the study by the same observer.
Adoptive transfer studies. In order to obtain AMs for adoptive transfer studies, airways of naive WT or IRF5−/− mice were lavaged six times with 0.5 ml of ice-cold PBS via a tracheal cannula. AMs comprised >97% of the retrieved population as assessed by cytospin preparation and flow cytometric analysis, respectively. Cells were stained with the vibrant blue system (Life Technologies, Carlsbad, CA) as per the manufacturer’s instructions before transfer (200,000 cells/recipient in 100 μl sterile PBS) and >70% of transferred cells could be recovered 24 h later. Anti-IL-13 antibody was a gift from UCB Celltech (Slough, UK) 10 mg kg−1 was administered before transfer of AMs, intraperitoneally at day −2 and −4, respectively. An isotype-matched antibody was used as a control.
Histology. Paraffin-embedded sections (4 μm) of lungs (apical lobe) were stained with hematoxylin/eosin, periodic acid–Schiff, and Sirius Red. For assessment of inflammatory infiltrate, a semiquantitative scoring system was used to grade the size of lung infiltrates as previously described.33 Briefly, a score of 5 signified a large (>3 cells deep) widespread inflammatory infiltrate around the majority of vessels and bronchioles, and a score of 1 represented a small (≤2 cells deep) number of inflammatory foci. For mucus assessment, airways were categorized according to the abundance of periodic acid–Schiff-positive goblet cells and assigned numerical scores (0, <5% goblet cells; 1, 5–25%; 2, 25–50%; 3, 50–75%; 4, >75%). The sum of the airway scores from each lung was divided by the number of airways examined to obtain the histological goblet cell score (expressed as mucus score in arbitrary units). All scoring and measurements were performed blinded by the same observer on medium airways measuring between 150 and 250 μm in diameter. Epithelial cell height and thickness of the airway smooth muscle layer around medium-sized conducting airways were measured from paraffin sections. At least six measurements were made per airway with a minimum of six airways measured per section. Data presented are % thickening in comparison with PBS-treated WT control.
Overexpression of IRF5. Selected groups received a first-generation replication-deficient AdIRF5 (0.5 × 109 viral plaque-forming units in PBS) or a control AdC 2 days before commencing instillation of either HDM or PBS.
Quantification of chemokines and immunoglobulins. Chemokine levels were measured in lung homogenates (cardiac lobe, 50 mg ml−1). Paired antibodies for murine eotaxin-2 and IL-10 (R&D Systems, Abingdon, UK), tumor necrosis factor-α, IL-1β, IL-6, IL-13 (eBiosciences, San Diego, CA), and IFN-γ (BD Biosciences, San Jose, CA) were used in standardized sandwich enzyme-linked immunosorbent assays according to the manufacturer’s protocol. Paired antibodies for IgE, IgG1, and IgG2a (R&D Systems) were used to measure serum and lung antibody levels.
Quantification of total collagen. Total collagen was measured in lung tissue (cardiac lobe) by biochemical assay (Sircol collagen assay, Biocolor, Belfast, UK) and normalized for tissue weight (50 mg ml−1).
Flow cytometric analysis. Disaggregated lung (left lobe) or BAL cells were either stained without stimulation or stained following stimulation with 500 ng ml−1 of ionomycin and 50 ng ml−1 of phorbol 2-myristate 13-acetate in the presence of brefeldin (BD Pharmingen, San Diego, CA) or HDM (50 μg ml−1). Cells were washed and preincubated with serum or Fc Block (2.4G2) before surface staining with the following antibodies purchased (clones in brackets): Biolegend (San Diego, CA): F4/80 (BM8), CD68 (FA-11), ICOS (C3984A); eBioscience (San Diego, CA): IL-13 (ebio13A), IL-17 (ebio17B7), GR-1 (RB6-8C5), CD11c (N418), CD45 (30-F11), CD11b (M1/70), IFN-γ (XMG1.2), lineage cocktail (17A2, RA3-6B2, M1/70, TER-119, RB6-8C5); eBiosciences: Ly6c (AL21), CD4 (RM4-5), Siglec-F (E50-2440), and T1/ST2 (RMST2-33). Total macrophage populations were identified by CD68 or F4/80 staining. Labeled cells were acquired on a BD fluorescence-activated cell sorting LSR Fortessa (BD Biosciences) and further analyzed by using FlowJo (Treestar, Ashland, OR). Intranuclear staining for IRF5 was carried out using a Rabbit polyclonal antibody to IRF5 (Abcam, Cambridge, UK, catalog ab178899), or for detection of full-length IRF5 in overexpression studies, a Rabbit polyclonal antibody to IRF5 (Abcam, catalog ab175317), followed by Alexa Fluor 488 Goat Anti-Rabbit IgG antibody (Life Technologies). Surface staining was followed by fixation and then permeabilization to allow for intracellular or intranuclear staining.
Real-time PCR. Total RNA was extracted from 50 to 100 mg of lung tissue (azygous lobe) using a Qiagen RNeasy Mini Kit (Hilden, Germany). Total RNA (1 μg) was reverse transcribed into cDNA using a High Capacity cDNA Reverse Transcription Kit (Life Technologies) as per the manufacturer’s instructions. Real-time PCR reactions were performed using fast-qPCR mastermix (Life Technologies) on a Viaa-7 instrument (Life Technologies) with TaqMan primer sets for murine IL-4, IL-5, IL-13, eotaxin-2, fibronectin, collagen-1α1, Mucin 5B, Mucin 5ac, Arg-1, YM-1, iNOs, TGF-β1, TGF-β2, TGF-β3, GAPDH, or HPRT (Life Technologies), and gene expression was analyzed using the change-in-threshold ΔΔCt method, and fold changes in mRNA expressions for targeted genes were calculated relative to WT controls.
Statistical analysis. Data were analyzed using Prism 6 for Windows from GraphPad Software (La Jolla, CA) using Kruskal–Wallis or Mann–Whitney tests.
References
Chanez, P. et al. Severe asthma in adults: what are the important questions? J. Allergy Clin. Immunol. 119, 1337–1348 (2007).
Anderson, G.P. Interactions between corticosteroids and beta-adrenergic agonists in asthma disease induction, progression, and exacerbation. Am. J. Respir. Crit. Care Med. 161, S188–S196 (2000).
Krausgruber, T. et al. IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115, 4421–4430 (2010).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and T. Nat. Immunol. 12, 231–238 (2011).
Ryzhakov, G., Eames, H.L. & Udalova, I.A. Activation and function of interferon regulatory factor 5. J. Interferon Cytokine Res. 35, 71–78 (2014).
Lee, Y.H., Bae, S.-C., Choi, S.J., Ji, J.D. & Song, G.G. Associations between interferon regulatory factor 5 polymorphisms and rheumatoid arthritis: a meta-analysis. Mol. Biol. Rep. 40, 1791–1799 (2012).
Lessard, C.J. et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 45, 1284–1292 (2013).
Salloum, R. & Niewold, T.B. Interferon regulatory factors in human lupus pathogenesis. Transl. Res. 157, 326–331 (2011).
Gathungu, G., Zhang, C.K., Zhang, W. & Cho, J.H. A two-marker haplotype in the IRF5 gene is associated with inflammatory bowel disease in a North American cohort. Genes Immun. 13, 351–355 (2012).
Wang, C. et al. Evidence of association between interferon regulatory factor 5 gene polymorphisms and asthma. Gene 504, 220–225 (2012).
Dobbs, L.G. & Johnson, M.D. Alveolar epithelial transport in the adult lung. Respir. Physiol. Neurobiol. 159, 283–300 (2007).
Byrne, A.J., Mathie, S.A., Gregory, L.G. & Lloyd, C.M. Pulmonary macrophages: key players in the innate defence of the airways. Thorax 70, 1189–1196 (2015).
Hussell, T. & Bell, T.J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14, 81–93 (2014).
Mathie, S.A. et al. Alveolar macrophages are sentinels of murine pulmonary homeostasis following inhaled antigen challenge. Allergy 70, 80–89 (2015).
Bedoret, D. et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Invest. 119, 3723–3738 (2009).
Kim, H.Y. et al. Interleukin-17–producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 20, 54–61 (2013).
Fang, C.-M. et al. Unique contribution of IRF-5-Ikaros axis to the B-cell IgG2a response. Genes Immun. 13, 421–430 (2012).
Feng, D. et al. Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. Eur. J. Immunol. 42, 1477–1487 (2012).
Roy, M.G. et al. Muc5b is required for airway defence. Nature 505, 412–416 (2013).
Zsengellér, Z., Otake, K., Hossain, S.A., Berclaz, P.Y. & Trapnell, B.C. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J. Virol. 74, 9655–9667 (2000).
Kim, E.Y. et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 14, 633–640 (2008).
Wills-Karp, M. Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261 (1998).
Hancock, A., Armstrong, L., Gama, R. & Millar, A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am. J. Respir. Cell Mol. Biol. 18, 60–65 (2012).
Grünig, G. et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282, 2261–2263 (1998).
Shirey, K.A. et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R. Mucosal Immunol. 3, 291–300 (2010).
Kang, C.-M. et al. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am. J. Respir. Cell Mol. Biol. 33, 290–296 (2005).
Yang, Z. et al. Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of type 2 immunity. PLoS One 8, e59441–e59441 (2013).
Saigusa, R. et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. PNAS 112, 15136–15141 (2015).
Dalmas, E. et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat. Med. 21, 610–618 (2015).
Tourdot, S. et al. Respiratory syncytial virus infection provokes airway remodelling in allergen-exposed mice in absence of prior allergen sensitization. Clin. Exp. Allergy 38, 1016–1024 (2008).
Anthony, D. et al. SAA drives proinflammatory heterotypic macrophage differentiation in the lung via CSF-1R-dependent signaling. FASEB J. 28, 3867–3877 (2014).
Stouch, A.N. et al. IκB kinase activity drives fetal lung macrophage maturation along a non-M1/M2 paradigm. J. Immunol. 193, 1184–1193 (2014).
Saglani, S. et al. Pathophysiological features of asthma develop in parallel in house dust mite–exposed neonatal mice. Am. J. Respir. Cell Mol. Biol. 41, 281–289 (2009).
Acknowledgements
We thank Lorraine Lawrence and Cecilia Andersson for histological sectioning and staining, Gaelle Herledan and Tom Shea for aiding with colony maintenance, Robert Snelgrove for sharing cells, Alessandra Lanfrancotti for the preparation of AdC and AdIRF5, and Kemi Awonaya for tissue processing. This work was supported by the American Asthma Foundation Early Excellence Award to I.A.U. (AAF 11-0105) and Wellcome Trust Senior Fellowship in Basic Biomedical Sciences to C.M.L. (087618/Z/08/Z).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
A.J.B. conducted the experiments, analyzed the data, and wrote the manuscript; M.W., S.A.M., S.A.W., H.L.E. and D.S. performed some of the experimental work; I.A.U. and C.M.L. designed the study, supervised the project, and edited the article.
Supplementary Material is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Byrne, A., Weiss, M., Mathie, S. et al. A critical role for IRF5 in regulating allergic airway inflammation. Mucosal Immunol 10, 716–726 (2017). https://doi.org/10.1038/mi.2016.92
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.92
This article is cited by
-
Macrophage metabolic reprogramming during chronic lung disease
Mucosal Immunology (2021)
-
Reducing IRF5 expression attenuates colitis in mice, but impairs the clearance of intestinal pathogens
Mucosal Immunology (2019)
