
Abstract
Colitis-associated colorectal cancer (CAC) is the most serious complication of inflammatory bowel disease (IBD). Excessive complement activation has been shown to be involved in the pathogenesis of IBD. However, its role in the development of CAC is largely unknown. Here, using a CAC model induced by combined administration of azoxymethane (AOM) and dextran sulfate sodium (DSS), we demonstrated that complement activation was required for CAC pathogenesis. Deficiency in key components of complement (e.g., C3, C5, or C5a receptor) rendered tumor repression in mice subjected to AOM/DSS. Mechanistic investigation revealed that complement ablation dramatically reduced proinflammatory cytokine interleukin (IL)-1β levels in the colonic tissues that was mainly produced by infiltrating neutrophils. IL-1β promoted colon carcinogenesis by eliciting IL-17 response in intestinal myeloid cells. Furthermore, complement-activation product C5a represented a potent inducer for IL-1β in neutrophil, accounting for downregulation of IL-1β levels in the employed complement-deficient mice. Overall, our study proposes a protumorigenic role of complement in inflammation-related colorectal cancer and that the therapeutic strategies targeting complement may be beneficial for the treatment of CAC in clinic.
Similar content being viewed by others


Introduction
Chronic inflammation represents a major driving force for cancer development and progression in many organs.1 For example, in patients with inflammatory bowel disease (IBD), such as ulcerative colitis, the risk of colorectal cancer development is much higher than in the general population.2 Long-standing ulcerative colitis predisposes to development of colitis-associated cancer (CAC), the major cause of death in patients with ulcerative colitis.3 Although the molecular mechanisms underlying transition from colitis to colorectal cancer remain elusive, the development of CAC is linked to overproduction of proinflammatory cytokines by immune and nonimmune cells, such as tumor necrosis factor-α, interleukin (IL)-6, and IL-17A.4, 5, 6, 7 These mediators have potentials to activate nuclear factor (NF)-κB signaling, thus leading to cell proliferation, augmented angiogenesis, and inhibition of cell death. Of note, recently we and other groups have proposed a disease-promoting role of proinflammatory cytokine IL-1β, mainly produced by infiltrating neutrophils, in CAC pathogenesis.8, 9 The protumorigenic function of IL-1β is shown to be associated with massive infiltration of neutrophils and elevated levels of proinflammatory cytokines and chemokines.
Complement, consisting of >30 plasma proteins and glycoproteins as well as soluble or membrane-bound receptors, is thought to be a chief component of innate immunity in host defense. However, excessive complement activation has been involved in the pathogenesis of inflammatory diseases, such as IBD. The expression of cell-surface complement-inhibitory proteins has been found to be decreased in gut epithelium of patients with IBD.10, 11 Furthermore, mice deficient in the complement inhibitor decay-accelerating factor exhibited decreased protection from complement activity and increased susceptibility to dextran sulfate sodium (DSS)-induced colitis.12 In accord with this notion, ablation of C3, the central component of the complement cascade, has been shown to be protective against DSS-induced colitis.13 Complement activation product C5a has also been reported to play a pathogenic role in acute trinitrobenzene sulfonic acid or DSS-induced colitis, validated by a C5a receptor (C5aR) antagonist,14 neutralizing anti-C5a antibody,15 or C5aR-deficient rodents.16
Although the implication of complement in the pathogenesis of IBD is well established, its role in the progression from colitis to cancer is poorly understood. In this study, we demonstrate that complement deficiency significantly repressed tumor formation in azoxymethane (AOM)/DSS-induced CAC model. This effect is related to dampened intestinal IL-1β/IL-17A axis.
Results
Complement activation during the course of CAC
Complement has been previously shown to be implicated in IBD pathogenesis. However, the evidence on complement activation during CAC is still absent. To address this, we assessed the kinetics of several parameters of complement activation in total colonic tissues after the initiation of CAC induction. Complement-activated product C3a was increased significantly in the colons during CAC (Figure 1b). Accordingly, the expression of C3 and C3a receptor was also elevated (Supplementary Figure S1A, B online). Intriguingly, C5a production was increased dramatically after CAC induction, and peaked on day 40 (Figure 1c). The amounts of C5a protein at this time point in the inflamed colons reached over 10-fold compared with normal controls. On day 60 (CAC induction was accomplished), colonic C5a contents decreased drastically, but was still higher than normal controls (Figure 1c). As interaction between C5a and C5aR easily leads to internalization of C5a/C5aR complex,17 the drop of C5a level may be because of this effect. In line with this notion, gene expression of C5 and C5aR in the lesions of CAC was persistently increased during CAC (Supplementary Figure S1C,D). Furthermore, C3 deposit in the colonic mucosa was visible during CAC (Figure 1d). Overall, these data suggest the occurrence of complement activation during the course of CAC.

Complement activation in azoxymethane/dextran sulfate sodium (AOM/DSS)-induced colitis-associated cancer. (a) Schematic overview of colitis-associated cancer (CAC) regimen. (b, c) At the indicated time points after initiation of CAC induction, colons were homogenated and supernatants were prepared for detection of (b) C3a and (c) C5a protein levels. Each group consisted of 6–8 mice. (d) C3 deposition in the colons at the indicated time points after CAC was detected by immunohistochemistry. Scale bar=100 μm. The data were pooled from two independent experiments. Each group consists of 6 to 8 mice. *P<0.05, **P<0.01. D, day.
Repression of CAC growth in complement-deficient mice
To determine whether there is causal relationship between complement activation and CAC formation, we established AOM/DSS-induced CAC model in C3-knockout mice and wild-type (WT) littermates. Compared with WT mice, C3 ablation significantly reduced tumor load (Figure 2a–c). Consistent with macroscopic change, increased apoptosis of colonic epithelial cells (CECs) was visible in C3−/− mice compared with WT littermates (Figure 2d). Moreover, the proliferation of CECs was limited in C3−/− mice, as reflected by reduced number of Ki-67-positive CECs (Figure 2e). This may be because of reduced activation of STAT3 (signal transducer and activator of transcription 3) and NF-κB as well as lower contents of anti-apoptotic protein Bcl-XL in C3−/− CECs (Figure 2f). To further investigate the involvement of complement cascade in CAC, we evaluated tumor growth in C5−/− and C5aR−/− mice, respectively. C5 or C5aR knockout conferred protection against CAC when AOM/DSS were administrated, resembling the phenotype of C3−/− mice (Supplementary Figure 2A,B). This indicates that complement is required for the progression from colitis to cancer.

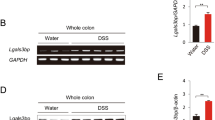
Complement deficiency reduces colitis-associated cancer (CAC) tumorigenesis. CAC was established in C3−/− mice and wild-type (WT) littermates according to the regimen as described in the Methods. The following parameters were evaluated on day 60. (a) Tumor number in colon and rectum was counted. (b) Tumor sizes were determined using Spot software for microscopic tumors or a caliper for macroscopic tumors. (c) Histologic examination with hematoxylin and eosin (H&E) staining was performed. Scale bar=500 μm (left panel); 200 μm (right panel). (d) Apoptosis of CECs was detected by TdT-mediated dNTP nick end labeling (TUNEL) methods. Quantitation of apoptotic cells is shown. Scale bar=100 μm. (e) Proliferation of CECs was detected by Ki-67-positive staining. Quantitation of proliferative cells is shown. Scale bar=100 μm. (f) Epithelial cells were isolated as described in the Methods and proteins were extracted. Signal transducer and activator of transcription 3 (STAT3) and nuclear factor (NF)-κBp65 subunit phosphorylation and B-cell lymphoma-extra large (Bcl-XL) contents were examined by western blotting. Changes in quantity of expression of these factors were determined by densitometric assays. The data were pooled from two or three independent experiments. Each group consists of 6 to 8 mice. *P<0.05; **P<0.01; ***P<0.001 vs. WT littermates. CEC, colonic epithelial cell; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
It has been generally accepted that a broad spectrum of cell types including epithelium and immune cells could synthesize and release complement components such as C3. In order to address the contribution of bone marrow or nonhematopoietic cell-derived C3 to CAC pathogenesis, we treated various bone marrow chimeric mice with the same combination of AOM and DSS. C3−/− mice transplanted with WT-derived bone marrow cells developed tumors at a similar level as WT mice transplanted with WT-derived bone marrow cells, higher than that in either WT or C3−/− mice transplanted with C3−/− mouse-derived bone marrow cells (Figure 3a). These observations suggest that bone marrow cell-derived C3, rather than non-bone marrow-derived cells, is crucial for tumor growth in this carcinogenesis model. Furthermore, because C5aR is expressed widely in immune cells and epithelial cells, to define whether C5aR-mediated signals on leukocytes or epithelium are critical for CAC development, we also performed bone marrow chimera experiments using C5aR−/− mice and WT littermates. C5aR−/− mice implanted with bone marrow cells from WT mice exhibited high CAC incidence that was comparable to WT mice with implantation of WT-derived bone marrow cells. However, less number of tumor nodules was observed in WT or C5aR−/− mice implanted with C5aR−/− mouse-derived bone marrow cells (Figure 3b). These data indicate C5a/C5aR signals on immune cells play a key role in CAC tumorigenesis.

Hematopoietic cell-derived complement is required for colitis-associated cancer (CAC) carcinogenesis. Bone marrow (BM) chimeric mice from (a) C3−/− and (b) C5aR−/− rodents were generated and subjected to azoxymethane/dextran sulfate sodium (AOM+DSS) treatment as described in the Methods. Colons were removed on day 60, and tumor number was counted. The bars represent the median of each group. Each group consists of 8 to 10 mice. ** P<0.01. WT, wild type.
Reduced IL-1β production and neutrophil infiltration in AOM/DSS-induced C3−/− mice
We next proceeded to detect the expression of a set of proinflammatory cytokines, well known to have a protumor role in CAC formation, in the lesions of C3−/− and WT mice subjected to AOM/DSS. Compared with WT controls, C3 deficiency dramatically reduced colonic IL-1β contents (Figure 4a). Intriguingly, other two putatively protumorigenic cytokines IL-6 and tumor necrosis factor-α were not disturbed in C3−/− mice (Supplementary Figure S3). As our previous study has identified neutrophil as the main producer of IL-1β in CAC milieu,8 we detected its expression in infiltrating neutrophil isolated from C3−/− and WT mice. Again, infiltrating neutrophil was verified as dominant cellular source of IL-1β (data not shown). As expected, IL-1β expression was decreased remarkably in C3−/− neutrophils (Figure 4b). Moreover, the number of infiltrating neutrophils in the lesions was significantly reduced in C3−/− mice, confirmed by immunohistochemistry (Figure 4c) and flow cytometry (Supplementary Figure S4). These observations indicate that tumor suppression in C3−/− mice may be partly attributed to reduction in IL-1β production in neutrophils and decreased recruitment of this polymorphonuclear population into lesions. This conception is consistent with the conclusion that neutrophil promotes the development of CAC by producing IL-1β, drawn by our previous study.8 To further address the causal role of IL-1β in this system, we injected recombinant IL-1β protein into C3−/− mice followed by administration of AOM and DSS. Indeed, transfusion of exogenous IL-1β resulted in enhanced and sustained intestinal production of IL-1β (Supplementary Figure S5). Importantly, this administration led to tumor recurrence in C3−/− mice, comparable to AOM/DSS-treated WT littermates (Figure 4d). Based on these observations, it is likely that tumor repression in C3−/− mice arises from downregulated expression of protumor cytokine IL-1β in the intestine.

Complement C3 ablation reduces interleukin (IL)-1β expression in infiltrating neutrophils. (a) Colitis-associated cancer (CAC) was induced in C3−/− mice and wild-type (WT) littermates according to the regimen as described in the Methods. On day 60, after initiating CAC induction, colon explants were cultured ex vivo for 24 h. IL-1β contents in the supernatants were determined by enzyme-linked immunosorbant assay (ELISA). (b) Neutrophils (Gr-1hiCD11b+) in lamina propria were isolated and IL-1β expression was examined by real-time reverse-transcriptase–PCR (RT-PCR). (c) Neutrophil infiltration in tumor tissues was detected by staining with anti-Gr-1 antibody. Scale bar=500 μm (upper) and 100 μm (lower). Right: summarized results. (d) C3−/− mice were injected intraperitoneally with recombinant mouse IL-1β (rIL-1β; 1 μg per mouse) or phosphate-buffered saline (PBS) every other day during CAC induction. On day 60, colons were dissected and tumor number was counted. Each group consists of 6 to 10 mice. *P<0.05; **P<0.01; ***P<0.001 vs. WT littermates. HPF, high-power field.
Dampened IL-1β/IL-17A axis in AOM/DSS-induced C3−/− mice
IL-1β has been reported to act as inducer for IL-17A response in the intestine.18, 19 The latter is well recognized as a key player in promoting CAC development.7 Thus, we speculate that tumor repression in C3−/− mice may be because of suppression of IL-1β-mediated IL-17A response in CAC niche. To address this hypothesis, we detected IL-17A level in the lesion of CAC-suffering C3−/− and WT mice. When compared with WT mice, IL-17A protein levels in the affected colon of C3−/− mice were dramatically reduced (Figure 5a). We also examined the colonic expression of other members of IL-17 family. IL-23, IL-17F, and IL-21 expression was not affected, whereas IL-22 level was also decreased significantly in AOM/DSS-treated C3−/− mice (Supplementary Figure S3). These observations indicate that intestinal IL-17A response was actively dampened in the absence of C3. To dissect the cellular source of IL-17A in CAC microenvironment, we separated lamina propria (LP) immune cells into four subpopulations on the basis of Gr-1 and CD11b staining and their IL-17A mRNA levels were measured. The results showed that IL-17A resided mainly in CD11b-positive myeloid cells including Gr-1hiCD11b+ cells (neutrophil) and Gr-1low/−CD11b+ cells (mononuclear phagocytes, namely macrophage and dendritic cells) and the abundance of this cytokine was relatively higher in neutrophil than that in mononuclear phagocytes (Figure 5b). We also analyzed the distribution of IL-17A expression in immune cell subsets by flow cytometry and confirmed CD11b+ myeloid cells as the main producer of this cytokine (Supplementary Figure S6A). Of importance, C3 deficiency resulted in significant downregulation of IL-17A expression in CD11b+ myeloid cells (Figure 5c and Supplementary Figure S6B).

Dysregulated intestinal interleukin (IL)-1β/IL-17A axis in the absence of complement C3. (a) On day 60 following initiation of colitis-associated cancer (CAC) induction, colon explants were cultured ex vivo for 24 h. IL-17A contents in the supernatants were determined by enzyme-linked immunosorbant assay (ELISA). (b) Immune cells in the lamina propria (LP) of CAC-bearing or naive mice were sorted by staining for Gr-1 and CD11b and divided into four subpopulations (left panel), Gr-1−CD11b−, Gr-1hiCD11b+, Gr-1+CD11b−, and Gr-1low/−CD11b+, and then IL-17A mRNA levels in these cells were examined by real-time reverse-transcriptase–PCR (RT-PCR). (c) CD11b+ myeloid cells in LP of C3−/− and wild-type (WT) mice subjected to azoxymethane/dextran sulfate sodium (AOM/DSS) were isolated and IL-17A mRNA expression was determined by real-time RT-PCR. (d) AOM/DSS-induced mice were treated with IL-1 receptor antagonist (IL-1Ra) or phosphate-buffered saline (PBS) according to the regimen as described in the Methods. On day 60, CD11b+ myeloid cells in LP were sorted and IL-17A mRNA expression was determined by real-time RT-PCR. (e) CD11b+ myeloid cells sorted from the colon LP of naive WT mice were stimulated with recombinant IL-1β (20 ng ml−1) alone or plus IL-23 (100 ng ml−1) for 24 h and then IL-17A expression was measured by real-time RT-PCR (left panel) or ELISA (right panel). (f) CD11b+ myeloid cells were sorted from the colon LP of naive WT mice and cocultured with neutrophil from the lesions of AOM/DSS-induced C3−/− and WT mice or naive WT mice for 24 h with or without IL-1Ra (2 μg ml−1) and then IL-17A levels in the supernatants were measured by ELISA. The data are pooled from two or three independent experiments. Each group consists of 5 to 6 mice. *P<0.05; **P<0.01; ***P<0.001 vs. WT littermates or controls. neu, neutrophil; US, untreated.
To assess whether IL-17A acted downstream of IL-1 signaling, we used IL-1 receptor antagonist (IL-1Ra) to block IL-1β bioactivity in vivo. As shown in our previous study,8 long-term treatment of IL-1Ra dramatically decreased the number of tumor nodules in the colon (data not shown). Next, we fractionated CD11b+ myeloid cells from the LP of mice with or without IL-1Ra treatment and detected IL-17A mRNA expression in this population. As shown in Figure 5d, IL-1Ra injection led to a significant reduction in IL-17A expression by CD11b+ myeloid cells, indicating that IL-1 signaling was required for IL-17A production by intestine-resident immune cells during CAC induction in vivo. To evaluate a direct effect of IL-1β on IL-17A production, intestinal CD11b+ myeloid cells were prepared from naive mice and exposed to exogenous IL-1β stimulation or combined with IL-23. The results showed that IL-1β challenge alone was sufficient to elicit the synthesis of IL-17A mRNA and protein (Figure 5e). Addition of IL-23 amplified IL-1β-driven IL-17 secretion in myeloid cells (Figure 5e). Furthermore, in the co-culture experiments, neutrophils isolated from the LP of CAC-bearing WT mice, but not naive or C3−/− mice, enabled to induce IL-17A production in CD11b+ myeloid cells and this effect was abrogated by addition of IL-1Ra to the medium (Figure 5f). Consistent with this observation, overexpression of IL-1β in vivo by injection of recombinant IL-1β into C3−/− mice efficiently augmented IL-17A expression in CD11b+ myeloid cells in the LP (Supplementary Figure S7). These results suggest that infiltrating neutrophils produce a high level of IL-1β and induces intestinal CD11b+ myeloid cells to express IL-17A via an autocrine/paracrine manner. Therefore, C3 deficiency blunted IL-1β/IL-17A axis in CAC milieu that may account for repression of CAC growth in the absence of C3.
C5a-elicited IL-1β expression by neutrophil
As described above, once the CAC model was established, the phenotype of C5aR−/− mice resembled that of C3- or C5-deficient mice (Figure 2). This finding led us to speculate that C5a/C5aR signals mediate the tumor-promoting effects of complement cascade during intestinal inflammation. To address this notion, neutrophils were isolated and stimulated by exogenous C5a. Indeed, C5a alone was sufficient to elicit robust production of IL-1β (Figure 6a). This effect was impaired by addition of a neutralizing anti-C5aR antibody or replaced by C5aR−/− neutrophil. In accord with this, neutrophils from AOM/DSS-induced C5aR−/− mice produced less amounts of IL-1β than those from WT mice (Figure 6b). Furthermore, although lipopolysaccharide challenge was sufficient to drive IL-1β release in neutrophil, C5a potently amplified this effect (Figure 6c), indicating synergistic actions of commensal flora-derived lipopolysaccharide and C5a on IL-1β production in the context of CAC. Thus, complement activation represents a key event for triggering the production of proinflammatory IL-1β during intestinal inflammation, thereby shaping tumor-fostering microenvironments. As it has been reported that MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase) signaling pathway is indispensible for C5a-mediated effector response during acute and chronic inflammation,20 we defined the role of MAPK/ERK signal in IL-1β release upon C5a stimulation. The results showed that exposure to C5a virtually led to phosphorylation of MAPK/ERK and activation of NF-κB in neutrophil in a time- and dose-dependent manner (Figure 6d). More importantly, blockade of MAPK/ERK or NF-κB signaling pathway by the addition of appropriate inhibitors drastically suppressed IL-1β expression in C5a-stimulated neutrophil (Figure 6e). These data indicate a nonredundant role of MAPK/ERK signaling pathway in C5a-elicted IL-1β production. Furthermore, in order to validate the capacity of C5a-exposed neutrophil to drive IL-17A expression in myeloid cells, intestinal CD11b+ myeloid cells were isolated and cocultured with neutrophils and C5a was added. C5a-challenged neutrophils actively drove myeloid cells to produce IL-17A (Figure 6f). Of note, this appeared not to be a direct effect of C5a on myeloid cells, as coculture of C5aR−/− myeloid cells with C5a-exposed neutrophils did not affect IL-17A release (Figure 6f). Therefore, C5a, a key component of complement cascade, potently drives neutrophil to produce proinflammatory cytokine IL-1β that drives protumorigenic IL-17A response in intestinal myeloid cells.

C5a acts as regulator for intestinal interleukin (IL)-1β/IL-17A axis. (a) Neutrophils from naive C5aR−/− and wild-type (WT) mice were isolated and stimulated with recombinant C5a (5 nM) for 6 h in the presence or absence of a neutralizing anti-C5a antibody (2 μg ml−1). IL-1β levels in the supernatants were examined by enzyme-linked immunosorbant assay (ELISA). (b) Colitis-associated cancer (CAC) was induced in C5aR−/− and WT mice according to the regimen as described in the Methods. On day 60, neutrophils infiltrating in the lesions were sorted and IL-1β mRNA expression was measured by real-time reverse-transcriptase–PCR (RT-PCR). (c) Naive neutrophils were stimulated by lipopolysaccharide (LPS; 10 ng ml−1) in the presence or absence of recombinant C5a (50 nM) for 6 h. IL-1β levels in the supernatants were examined by ELISA. (d) Neutrophils from naive WT mice were stimulated with C5a at the indicated concentrations for 5 min or at the dose of 50 nM for the indicated time respectively. Mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase-1/2 (ERK1/2) and nuclear factor (NF)-κBp65 subunit phosphorylation was detected by western blotting. (e) Neutrophils were stimulated with C5a (50 nM) in the presence or absence of ERK inhibitor (U0126, 10 μm) or NF-κB inhibitor (Bay11-7082, 20 μm) for 6 h. IL-1β levels in the supernatants were examined by ELISA. (f) Neutrophils from naive WT mice were stimulated with C5a (50 nM) and cocultured with intestinal CD11b+ myeloid cells from C5aR−/− and WT mice for 12 h. IL-17A levels in the supernatants were measured by ELISA. The data were pooled from three independent experiments. Each group consists of 6 to 8 mice.**P<0.01; ***P<0.001 vs. WT littermates. neu, neutrophil; US, untreated.
Discussion
It is well established that complement system is definitely involved in the pathophysiological process of IBD. Aberrant or excessive complement activation contributes to IBD pathogenesis. Its role in transition from colitis to colorectal cancer, however, is not reported to date. In this study, we provide evidence for the first time that complement activation contributes to the development of CAC. Complement split protein C5a represents a key component for CAC formation by triggering robust production of proinflammatory IL-1β in infiltrating neutrophils, thereby eliciting protumor IL-17A response in intestinal myeloid cells. It is known that C5a can utilize different mechanisms to facilitate tumor growth in several types of solid tumors including: recruitment of myeloid-derived suppressor cells into the surroudings of neoplasia;21, 22 inducing endothelial cell chemotaxis and neovascularization;23 and directly promoting tumor growth in cancer cells expressing C5aR.24 The C5a signals on epithelial cells seem to be dispensable for CAC tumorigenesis as transfusion of WT mice-derived bone marrow cells into C5aR−/− recipients renders tumor recurrence. Whether other functions of C5a are available in our model need further investigation. Overall, our present study proposes a novel function of C5a on intestinal IL-1β-IL-17A axis in tumor niche that is critical for CAC development.
IL-1β is an important mediator for promoting chronic inflammation and malignant growth and progression.25, 26, 27 In IBD, very high levels of IL-1β in the intestine were observed in patients and rodents.28, 29, 30, 31 Treatment with IL-1-blocking agents has been successful in ameliorating acute models of intestinal injury and inflammation.32 Furthermore, different genetic lesions associated with IBD development in animal models are associated with increased IL-1β.33 Recently, our work and others have demonstrated that inflammasome-regulated IL-1β release is a key event for the progression from colitis to cancer.8, 9 We found that complement deficiency led to significant decrease of IL-1β in the intestine. Overproduction of this cytokine in vivo rendered tumor recurrence in complement-deficient mice when AOM/DSS was administrated. These data support that complement favors the development of CAC by regulating IL-1β expression. Furthermore, consistent with our previous study,8 the dissection of cellular source showed that neutrophils infiltrating into colon lesions were main producer for IL-1β. Thus, we concluded that complement activation components (e.g., anaphylatoxin C5a and C3a) regulated IL-1β production by neutrophils, thereby promoting CAC development. Of note, C5a enabled to upregulate IL-1β mRNA levels and promote the release of mature IL-1β (Figure 6a; G-J Chen, unpublished data). Thus, C5a-elicited IL-1β production may be attributed to enhanced IL-1β transcription and/or pro-IL-1β processing. The details need further investigations. Besides regulating cytokine production, C5a is also a strong chemoattractant for immune cells including neutrophils and delays apoptosis of neutrophil. These effects are in line with the data that complement deficiency resulted in reduction in the number of infiltrating neutrophils in CAC-bearing mice.
IL-1β is a proinflammatory cytokine with a wide range of systemic and local biological functions. Recent studies have demonstrated that intestinal IL-1β is pivotal for accumulation of IL-17A-secreting innate and adaptive immmune cells in the intestine under steady state and during chronic inflammation.18, 19 Accordingly, we provide evidence that complement deficiency blunts IL-1β-elicited IL-17A response in intestinal myeloid cells. Considering a defined tumor-promoting role of IL-17A in CAC pathogenesis,7 dampened IL-17A response may account for tumor suppression in complement-deficient mice that is related to alteration of proliferation/apoptosis by downregulation of IL-17A-mediated STAT3/NF-κB signaling pathway. Intriguingly, IL-17A distribution in immune cell populations seems to be dependent on intestinal inflammation models used. In Helicobacter hepaticus-infected model, intestinal IL-17A is predominantly secreted by T helper type 17 cells and innate lymphoid cells.18 However, enterotoxigenic Bacteroides fragilis triggers colitis and induces colonic tumors by enhancing IL-17A response in CD4+αβTCR+ T cells and CD4−CD8−γδTCR+ T cells in the intestine.34 In our study, we demonstrated high abundance of IL-17A in CD11b+ myeloid cells including neutrophils and mononuclear phagocytes during AOM/DSS-induced inflammation, in line with the conception that innate immune system plays a dominant role in this model.35 Notably, a recent study proposed the importance of IL-17-producing γδT cells in promoting colorectal tumor growth.36 Thus, it is plausible that, in the context of colorectal cancer, the pattern of IL-17 distribution in immune cells is regulated by local microenvironment and myeloid IL-17 response is crucial for tumor growth and progression in inflammation-driven colorectal tumorigenesis. Furthermore, neutrophil and macrophage represent the sources of IL-17A, identified previously in many diseases, for example arthritis,37 asthma,38 kidney ischemia/reperfusion injury,39 antineutrophil cytoplasmic antibody-associated vasculitis,40 and sepsis.41 It is noteworthy that a direct effect of C5a on IL-17A release by CD11b+ myeloid cells should be ruled out because C5a stimulation antagonizes rather than induces IL-17A production by macrophage (G-J Chen, unpublished data). Thus, neutrophil/IL-1β-myeloid cells/IL-17A axis is critical for CAC development, and this pathway appears to be compromised specifically in the absence of complement. In addition, we found that complement deficiency also led to decreased expression of IL-22 in the lesions (Supplementary Figure S3). This effect was reversed by administration of recombinant IL-1β (G-J Chen, unpublished data). Given that the importance of IL-22 in colitis an colon cancer has been highlighted recently,42, 43 the details in regulation of IL-22 as well as IL-17A by IL-1β are currently under investigation.
Our study proposed a schematic model of complement function on CAC carcinogenesis as follows: oral administration of DSS leads to aberrant complement activation. Complement-activated products (particularly C5a) trigger neutrophils to produce large amounts of proinflammatory cytokine IL-1β. IL-1β potently induces a strong IL-17A response in intestine-residing CD11b+ myeloid cells via autocrine and/or paracrine manner. IL-17A promotes hyperproliferation of CEC through activating STAT3/NF-κB-dependent pathway, thereby enhancing its tumorigenic capacity. Notably, one of the pathological functions of IL-17A in inflammatory diseases is well known to act on accumulation of neutrophils at the site of inflammation.44 Thus, it is reasonable that IL-17A has a positive feedback on the recruitment of neutrophils into the lesions and augments the vicious circuits of neutrophil/IL-1β-CD11b+ myeloid cells/IL-17A and ultimately perpetuates the onset of CAC. In agreement with this conception, complement ablation dramatically attenuated intestinal injury in mice suffering from colitis as well as reduced IL-1β and IL-17 expression in the lesions (unpublished data (G-J Chen)), indicating that CAC suppression is mainly ascribed to decreased intestinal inflammation and downregulation of proinflammatory cytokines in complement-deficient mice. Intriguingly, recent studies have demonstrated the involvement of IL-17 response in colorectal cancer development.36, 45 Whether IL-1β/IL-17 axis is also critical for spontaneous colorectal tumorigenesis is worthy to be addressed in the future. Overall, our findings propose the importance of complement in CAC tumorigenesis and shed light on therapeutic potentials of complement-targeted drugs (e.g., compounds and antibody to C5a) in ulcerative colitis-related colorectal cancer in clinic.
Methods
Mice. B6 (C57BL/6 J), C3−/−, C5−/−, and C5aR−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in separated cages as well as bred as independent colonies in specific pathogen-free conditions on an alternating light/dark cycle. All animal experiments were performed in accordance with international guidelines for the care and use of laboratory animals and approved by the Animal Ethics Committee of the Institute of Basic Medical Sciences.
Acute colitis and CAC induction. Acute colitis was induced in mice by daily drinking water containing 3% DSS (molecular weight 36,000–50,000; MP Biochemicals, Santa Ana, CA) for 5 days.
CAC was induced according to classical protocols as described previously,46 with mild modification (Figure 1a). In brief, on day 0, mice were injected intraperitoneally with AOM (10 mg kg−1; Sigma-Aldrich, St. Louis, MO) and maintained on regular diet and water for 5 days. Mice then received water with 1.5% DSS for 1 week. After this, mice were maintained on regular water for 2 weeks and subjected to two more DSS treatment cycles. On day 60, mice were killed. Macroscopic tumors were counted. The clinical course of disease was followed daily by measurement of body weight and monitoring for signs of rectal bleeding or diarrhea.
Histological examination and immunohistochemistry. The protocols on immunohistochemistry are described in Supplementary Materials and Methods.
Detection and quantitation of apoptotic cells. Experiments were carried out as described in Supplementary Materials and Methods.
Isolation of colonic epithelial cells and LP immune cell subsets. Experiments were performed according to the protocol described in detail in our previous study.8
Immunoblotting. Standard procedures for immunoblotting are described in Supplementary Materials and Methods.
Colon homogenates. The experiments were done as described in Supplementary Materials and Methods.
Whole colon culture. The protocol is described in Supplementary Materials and Methods.
Cytokine analysis. Cytokines (IL-1β, IL-17A, IL-6, tumor necrosis factor-α, and IL-23) and anaphylatoxin C3a and C5a were examined by ELISA kits obtained from R&D Systems (Minneapolis, MN), according to the manufacturer’s instructions.
Generation of bone marrow chimeric mice. The protocol is described in Supplementary Materials and Methods.
IL-1β and IL-1 receptor antagonist administration in vivo. During CAC induction (from day 0 to day 60), C3−/− mice were treated every other day with either phosphate-buffered saline or 1 μg per mouse of recombinant mouse IL-1β (Peprotech, Rocky Hill, NJ) intraperitoneally, and then intestinal tissue was harvested for analysis. To block IL-1 signaling, mice were injected intraperitoneally with recombinant human IL-1 receptor antagonist (IL-1Ra), kindly provided by Haiyan Hong (Beijing Proteomics Research Center, Beijing, China), at a dose of 0.5 mg per mouse on days 1, 3, and 5 of every cycle of DSS drinking.
Isolation of spleen cells, mouse LP immune cells, and in vitro treatment. The protocol is described in Supplementary Materials and Methods.
Flow cytometry. The protocol is described in Supplementary Materials and Methods.
Quantitative reverse transcriptase–PCR. Experiments were carried out as described in Supplementary Materials and Methods. The primers for real time are listed in Supplementary Table S1.
Statistical analysis. Data are presented as mean±s.d. Nonparametric test and Student’s t-test (two tailed) was used to determine significance, with P<0.05 considered significant. Statistics were performed using SPSS 10.0 for Macintosh, and graphs were made on Deltagraph (SPSS, Chicago, IL).
References
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Itzkowitz, S.H. & Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G7–G17 (2004).
Eaden, J.A., Abrams, K.R. & Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48, 526–535 (2001).
Popivanova, B.K. et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570 (2008).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Matsumoto, S. et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J. Immunol. 184, 1543–1551 (2010).
Hyun, Y.S. et al. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 33, 931–936 (2012).
Wang, Y. et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 7, 1106–1115 (2014).
Zhang, J., Fu, S., Sun, S., Li, Z. & Guo, B. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol. 7, 1139–1150 (2014).
Scheinin, T. et al. Decreased expression of protectin (CD59) in gut epithelium in ulcerative colitis and Crohn's disease. Hum. Pathol. 30, 1427–1430 (1999).
Berstad, A.E. & Brandtzaeg, P. Expression of cell membrane complement regulatory glycoproteins along the normal and diseased human gastrointestinal tract. Gut 42, 522–529 (1998).
Lin, F., Spencer, D., Hatala, D.A., Levine, A.D. & Medof, M.E. Decay-accelerating factor deficiency increases susceptibility to dextran sulfate sodium-induced colitis: role for complement in inflammatory bowel disease. J. Immunol. 172, 3836–3841 (2004).
Lu, F., Fernandes, S.M. & Davis, A.E. 3rd The role of the complement and contact systems in the dextran sulfate sodium-induced colitis model: the effect of C1 inhibitor in inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G878–G883 (2010).
Woodruff, T.M. et al. A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J. Immunol. 171, 5514–5520 (2003).
Chen, G. et al. Blockade of complement activation product C5a activity using specific antibody attenuates intestinal damage in trinitrobenzene sulfonic acid induced model of colitis. Lab. Invest. 91, 472–483 (2011).
Johswich, K. et al. Role of the C5a receptor (C5aR) in acute and chronic dextran sulfate-induced models of inflammatory bowel disease. Inflam. Bowel Dis. 15, 1812–1823 (2009).
Guo, R.F. et al. Neutrophil C5a receptor and the outcome in a rat model of sepsis. FASEB J. 17, 1889–1891 (2003).
Coccia, M. et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 209, 1595–1609 (2012).
Shaw, M.H., Kamada, N., Kim, Y.G. & Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 209, 251–258 (2012).
Issuree, P.D., Pushparaj, P.N., Pervaiz, S. & Melendez, A.J. Resveratrol attenuates C5a-induced inflammatory responses in vitro and in vivo by inhibiting phospholipase D and sphingosine kinase activities. FASEB J. 23, 2412–2424 (2009).
Markiewski, M.M. et al. Modulation of the antitumor immune response by complement. Nat. Immunol. 9, 1225–1235 (2008).
Corrales, L. et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 189, 4674–4683 (2012).
Nozaki, M. et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 103, 2328–2333 (2006).
Cao, Q., McIsaac, S.M. & Stadnyk, A.W. Human colonic epithelial cells detect and respond to C5a via apically expressed C5aR through the ERK pathway. American journal of physiology. Cell Physiol. 302, C1731–C1740 (2012).
Schauer, I.G. et al. Interleukin-1beta promotes ovarian tumorigenesis through a p53/NF-kappaB-mediated inflammatory response in stromal fibroblasts. Neoplasia 15, 409–420 (2013).
Liu, Q. et al. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 73, 3297–3305 (2013).
Huang, Q. et al. IL-1beta-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 13, 18 (2014).
Reinecker, H.C. et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin. Exp. Immunol. 94, 174–181 (1993).
McAlindon, M.E., Hawkey, C.J. & Mahida, Y.R. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 42, 214–219 (1998).
Okayasu, I. et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 (1990).
McCall, R.D. et al. Tissue interleukin 1 and interleukin-1 receptor antagonist expression in enterocolitis in resistant and susceptible rats. Gastroenterology 106, 960–972 (1994).
Siegmund, B., Lehr, H.A., Fantuzzi, G. & Dinarello, C.A. IL-1beta-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA 98, 13249–13254 (2001).
Maeda, S. et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science 307, 734–738 (2005).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17T cell responses. Nat. Med. 15, 1016–1022 (2009).
Dieleman, L.A. et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107, 1643–1652 (1994).
Wu, P. et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800 (2014).
Katayama, M. et al. Neutrophils are essential as a source of IL-17 in the effector phase of arthritis. PLoS One 8, e62231 (2013).
Ferretti, S., Bonneau, O., Dubois, G.R., Jones, C.E. & Trifilieff, A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170, 2106–2112 (2003).
Li, L. et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest. 120, 331–342 (2010).
Hoshino, A. et al. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J. Autoimmun. 31, 79–89 (2008).
Bosmann, M., Sarma, J.V., Atefi, G., Zetoune, F.S. & Ward, P.A. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. 26, 1640–1651 (2012).
Kirchberger, S. et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 210, 917–931 (2013).
Longman, R.S. et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211, 1571–1583 (2014).
Kolls, J.K. & Linden, A. Interleukin-17 family members and inflammation. Immunity 21, 467–476 (2004).
Grivennikov, S.I. et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 (2012).
Wirtz, S., Neufert, C., Weigmann, B. & Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546 (2007).
Acknowledgements
We thank Dr Haiyan Hong (Beijing Proteomics Research Center) for providing the IL-1Ra. This work was supported by grants from the National Key Basic Research Program of China (2013CB530506), the National Natural Science Foundation of China (81472647, 81272320, and 81072475), Beijing Natural Science Foundation (7132151), and Service Industry Scientific Research of National Health and Family Planning Commission of China (2015SQ00192).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Ning, C., Li, YY., Wang, Y. et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1β/IL-17A axis. Mucosal Immunol 8, 1275–1284 (2015). https://doi.org/10.1038/mi.2015.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.18
This article is cited by
-
Promotion of the inflammatory response in mid colon of complement component 3 knockout mice
Scientific Reports (2022)
-
Identification of glycophorin C as a prognostic marker for human breast cancer using bioinformatic analysis
Network Modeling Analysis in Health Informatics and Bioinformatics (2022)
-
Dynamic roles of inflammasomes in inflammatory tumor microenvironment
npj Precision Oncology (2021)
-
Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression
Nature Cancer (2021)
-
Integrated omics profiling of dextran sodium sulfate-induced colitic mice supplemented with Wolfberry (Lycium barbarum)
npj Science of Food (2020)
