
Abstract
Chronic obstructive pulmonary disease (COPD) is a life-threatening inflammatory respiratory disorder, often induced by cigarette smoke (CS) exposure. The development of effective therapies is impaired by a lack of understanding of the underlining mechanisms. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytokine with inflammatory and apoptotic properties. We interrogated a mouse model of CS-induced experimental COPD and human tissues to identify a novel role for TRAIL in COPD pathogenesis. CS exposure of wild-type mice increased TRAIL and its receptor messenger RNA (mRNA) expression and protein levels, as well as the number of TRAIL+CD11b+ monocytes in the lung. TRAIL and its receptor mRNA were also increased in human COPD. CS-exposed TRAIL-deficient mice had decreased pulmonary inflammation, pro-inflammatory mediators, emphysema-like alveolar enlargement, and improved lung function. TRAIL-deficient mice also developed spontaneous small airway changes with increased epithelial cell thickness and collagen deposition, independent of CS exposure. Importantly, therapeutic neutralization of TRAIL, after the establishment of early-stage experimental COPD, reduced pulmonary inflammation, emphysema-like alveolar enlargement, and small airway changes. These data provide further evidence for TRAIL being a pivotal inflammatory factor in respiratory diseases, and the first preclinical evidence to suggest that therapeutic agents that target TRAIL may be effective in COPD therapy.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of morbidity and death worldwide and imparts a major socioeconomic burden.1 It is a complex heterogeneous disease characterized by chronic pulmonary inflammation, airway remodeling and emphysema, that are associated with progressive lung function decline.2 It is primarily caused by cigarette smoke (CS) exposure in Western countries but additional factors are also important in other areas.3 Once induced the patient’s condition continues to deteriorate often even after smoking cessation.4 Glucocorticoids, anticholinergic agents, and long-acting muscarinic antagonists are the current mainstay therapies for COPD. However, these agents are poorly effective and do not modify the inducing factors or halt the progression of disease even at high doses.5 The lack of effective treatments for COPD is largely due to the poor understanding of the underlying mechanisms of disease pathogenesis.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), also known as tumor necrosis factor superfamily member 10 (TNFSF10), is a cytokine that induces both inflammation and apoptosis.6 TRAIL is expressed by a variety of cells including epithelial cells, monocytes/macrophages, neutrophils, dendritic cells, and T cells.7, 8, 9, 10, 11 Four cell surface receptors have been identified for TRAIL; death receptor 4 (DR4, also known as TRAIL-R1, which is not expressed in mice), DR5 (TRAIL-R2), decoy receptor 1 (DcR1; TRAIL-R3), and DcR2 (TRAIL-R4).12, 13, 14 Both DR4 and DR5 possess an intracellular death domain that induces apoptosis.12, 13 In contrast, DcR1 and DcR2 lack a functional death domain and, therefore, act as decoy receptors.14 Emerging evidence from us, and others, implicate TRAIL in chronic lung diseases such as asthma and pulmonary fibrosis.6, 7, 9, 15, 16 Experimental studies show that TRAIL promotes allergic airway disease (AAD) by increasing (C–C motif) ligand (CCL)20 production and myeloid dendritic cell (mDC) migration into the lungs resulting in increased inflammation and airway hyperresponsiveness.7 TRAIL-dependent signaling pathways are also critical for rhinovirus (RV)-induced AAD exacerbations.17 Furthermore, TRAIL promotes Chlamydia respiratory infection-induced pulmonary inflammation, airway hyperresponsiveness, and emphysema-like alveolar enlargement.6 It is also implicated in collagen deposition in a mouse model of ovalbumin-induced AAD,16 but conversely appears to protect against lung injury and fibrosis in some situations in mice.9 The role of TRAIL in CS-induced inflammation, airway remodeling, emphysema, impaired lung function, and the pathogenesis of COPD is unknown.
Here, we investigated the role of TRAIL using our recently established mouse model of chronic CS-induced experimental COPD that recapitulates the critical features of human disease.18, 19, 20, 21, 22, 23 TRAIL and its receptors were increased in the model and in lung samples from human COPD patients. The absence of TRAIL in deficient mice or the treatment of wild-type (WT) mice with an anti-TRAIL neutralizing antibody, significantly reduced the severity of experimental COPD. The mechanisms involved were investigated. This study is the first to characterize the role of TRAIL in COPD pathogenesis and identifies TRAIL as a potential therapeutic target for this disease.
Results
TRAIL mRNA expression and protein levels increase in CS-induced experimental COPD
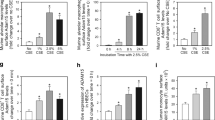
To investigate whether TRAIL expression is altered during the pathogenesis of COPD, we interrogated our established model of experimental COPD in mice.18, 19, 20, 21, 22, 23, 24 Chronic CS exposure for 8 weeks to induce experimental COPD significantly increased TRAIL messenger RNA (mRNA; Figure 1a,b) and protein levels (Figure 1c,d) in the airways and parenchyma, compared with control mice that breathed normal air, determined using real-time quantitative PCR (qPCR) and enzyme-linked immunosorbent (ELISA) assay. These data were supported by increases in TRAIL expression on small airway epithelial cells and parenchyma-associated inflammatory cells in histology sections in COPD groups detected by immunohistochemistry (Figure 1e and Supplementary Figure S1 online). CD11b+ monocytes were the predominant TRAIL-expressing cells in lung homogenates by flow cytometry (Figure 1f,g). Minimal changes were observed in the numbers of TRAIL+ neutrophils, gamma delta T (γδT) cells, natural killer T (NKT) cells or CD4+ and CD8+ T cells. Chronic CS exposure also increased DR5 mRNA expression in the airways (Figure 1h) but not parenchyma (Supplementary Figure S2a). DcR1 mRNA was increased in parenchyma (Figure 1i) but not airways and DcR2 expression was unaltered in both the airways and parenchyma (Supplementary Figure S2b–d).

Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor levels increase in cigarette smoke (CS)-induced experimental chronic obstructive pulmonary disease (COPD). Wild-type (WT) BALB/c mice were exposed to CS or normal air for eight weeks. TRAIL messenger RNA (mRNA) levels in blunt-dissected (a) airway and (b) parenchyma expressed as relative abundance to normal air-exposed controls. TRAIL protein levels in (c) airway and (d) parenchyma. (e) Immunohistochemistry for TRAIL protein in whole-lung sections (arrowheads indicate TRAIL+ epithelial cells in the small airways or inflammatory cells in the parenchyma). (f) Cell surface expression of TRAIL on inflammatory cell subsets in whole-lung homogenates determined by flow cytometry. (g) Representative flow cytometry histogram of TRAIL expression on CD45+F4/80+ CD11c−CD11b+ cells in lung homogenates from normal air- (red) and CS-exposed (cyan) WT mice. (h) Death receptor 5 and (i) decoy receptor 1 mRNA levels in airways or parenchyma. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. *P<0.05; **P<0.01; ****P<0.0001 compared with normal air-exposed controls.
TRAIL expression increases in human COPD
Given that TRAIL was increased in the airways and parenchyma in CS-induced experimental COPD, we next sought to determine whether the expression of TRAIL and its receptor mRNA was altered in equivalent tissues and cells from humans with COPD. TRAIL mRNA expression was assessed in pre-existing microarray data from COPD patients and non-COPD subjects.25, 26, 27 TRAIL expression was significantly increased ∼2.3-fold in airway epithelial brushings from patients with COPD compared with both nonsmokers and healthy smokers without COPD (Affymetrix Human Genome U133 Plus 2.0 Array, Accession: GSE5058,27 Figure 2a). TRAIL mRNA expression was also significantly increased approximately four-fold in the parenchyma of COPD patients compared with subjects without COPD (Affymetrix Human Exon 1.0 ST Array, Accession: GSE27597,25, 26 Figure 2b). The mRNA expression of human TRAIL receptors was also assessed in the same data. The TRAIL receptors DR4 (TNFRSF10A) and DR5 (TNFRSF10B), but not DcR1 (TNFRSF10C) or DcR2 (TNFRSF10D) were increased in epithelial brushings of COPD patients compared with nonsmokers and healthy smokers (Figure 2c–f). However, the expression of these receptors was not altered in the parenchyma (Supplementary Figure S3a–d). These data show increased TRAIL mRNA expression in the airways and parenchyma, and DR5 expression in the airways in both experimental and human COPD.

Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor messenger RNA (mRNA) levels increase in human chronic obstructive pulmonary disease (COPD). Parenchymal cores or airway epithelial cells were collected from human COPD patients. TRAIL mRNA expression was assessed by microarray profiling in (a) airway epithelial brushings from patients with the Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage I or II disease compared with nonsmokers (NS) and healthy smokers without COPD (smoker) and (b) parenchyma from non-COPD controls or patients with severe COPD. mRNA expression of TRAIL receptors (c) death receptor (DR)4, (d) DR5, (e) decoy receptor (DcR)1, and (f) DcR2 in airway epithelium from patients with early-stage, GOLDI and GOLDII stage COPD compared with NS and healthy smokers without COPD. The numbers in the figures represent the false discovery rate (FDR). *FDR vs. NS, #FDR vs. smokers.
Pulmonary inflammation is reduced in TRAIL-deficient mice exposed to CS
Given that TRAIL increased in experimental COPD, we next determined whether this cytokine has a role in CS-induced pulmonary inflammation. WT and TRAIL-deficient (Tnfsf10−/−) mice were exposed to CS to induce experimental COPD and pulmonary inflammation was assessed in bronchoalveolar lavage (BAL) by staining and differential inflammatory cell enumeration. CS exposure of WT mice significantly increased total leukocytes, macrophages, neutrophils, and lymphocytes in BAL compared with normal air-exposed WT controls (Figure 3a–d). In contrast, CS did not significantly increase total leukocytes or macrophages in Tnfsf10−/− mice, whereas neutrophils and lymphocytes were elevated compared with normal air-exposed Tnfsf10−/− controls. CS-exposed Tnfsf10−/− mice also had decreased total leukocytes and macrophages, but not neutrophils or lymphocytes, compared with CS-exposed WT controls (Figure 3a–d).

Pulmonary inflammation is reduced in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-deficient mice exposed to cigarette smoke (CS). Wild-type (WT) or TRAIL-deficient (Tnfsf10−/−) mice were exposed to CS or normal air for 8 weeks. (a) Total leukocytes, (b) macrophages, (c) neutrophils, and (d) lymphocytes were enumerated in May-Grunwald Giemsa-stained bronchoalveolar lavage (BAL). (e) The numbers of parenchymal inflammatory cells (arrowheads) were enumerated in periodic acid-Schiff-stained lung sections. (f) CD11b+ monocytes, (g) myeloid dendritic cells (mDCs) and (h) gamma delta T (γδT) cells were determined in single cell suspension of whole-lung homogenates by flow cytometry. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. NS, not significant. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001 compared with normal air-exposed WT or Tnfsf10−/− controls. #P<0.05; ##P<0.01; ###P<0.001 compared with CS-exposed WT controls.
Next, we assessed inflammatory cell numbers in the parenchyma by histology. CS exposure of WT mice significantly increased inflammatory cell numbers in the parenchyma compared with normal air-exposed WT controls (Figure 3e). CS exposure of Tnfsf10−/− mice also increased inflammatory cell numbers compared with normal air-exposed Tnfsf10−/−controls. However, CS-exposed Tnfsf10−/− mice had significantly reduced inflammatory cells compared with CS-exposed WT controls.
As CS-exposed Tnfsf10−/− mice had marked reductions in both BAL and parenchymal inflammatory cells, we next determined if there were any differences in the numbers of individual inflammatory cell types in the lung by flow cytometry (Table 1). CS exposure of WT mice increased the numbers of CD11b+ monocytes, mDCs, and γδT cells in lung compared with normal air-exposed WT controls (Figure 2f–h). CS exposure of Tnfsf10−/− mice also increased CD11b+ monocytes and mDCs, but not γδT cells compared with normal air-exposed Tnfsf10−/− controls. However, CS-exposed Tnfsf10−/− mice had reduced CD11b+ monocytes, mDCs and γδT cells compared with CS-exposed WT controls. There were no differences in the numbers of alveolar macrophages, plasmacytoid (p)DCs, natural killer T cells, CD4+ or CD8+ T cells between CS-exposed Tnfsf10−/− and WT mice (Supplementary Figure S4a–e).
Pro-inflammatory cytokine, chemokine, and COPD-related factor mRNA expression are reduced in TRAIL-deficient mice exposed to CS
Given that CS-exposed Tnfsf10−/− mice had decreased pulmonary inflammatory cells, we next assessed the mRNA expression of inflammatory cytokines, chemokines, and COPD-related factors in the lung. CS exposure of WT mice increased the mRNA expression of the cytokine tumor necrosis factor-α (TNF-α; Figure 4a), the chemokines CCL2, 3, 7, 12, and 20 (Figure 4b–f) and other COPD-related factors matrix metalloproteinase-12 (MMP-12) and serum amyloid A3 (SAA3, Figure 4g,h) compared with normal air-exposed WT controls. CS exposure of Tnfsf10−/− mice also increased the mRNA expression of these cytokines, chemokines, and COPD-related factors compared with normal air-exposed Tnfsf10−/− controls. However, CS-exposed Tnfsf10−/− mice had significantly reduced expression of all of these factors compared with CS-exposed WT mice. We also profiled other factors including interleukin (IL)-33, chemokine (C–X–C) ligand (CXCL)1 and 3, CCL4 and 22 and mucin 5ac (Supplementary Figure S5a–f). These were increased by CS but were not different between CS-exposed WT and Tnfsf10−/− mice.

Pro-inflammatory cytokine, chemokine, and chronic obstructive pulmonary disease (COPD)-related factor messenger RNA (mRNA) expression are reduced and NF-κB p65 activity is inhibited in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-deficient mice exposed to cigarette smoke (CS). Wild-type (WT) or TRAIL-deficient (Tnfsf10−/−) mice were exposed to CS or normal air for 8 weeks. (a) Tumor necrosis factor-α (TNF-α), (b) chemokine (C–C motif) ligand (CCL)2, (c) CCL3, (d) CCL7, (e) CCL12, (f) CCL20, (g) matrix metalloproteinase-12 (MMP-12), and (h) serum amyloid A3 (SAA3) mRNA expression was determined in whole-lung homogenates by qPCR. (i) NF-κB p65 activity in whole-lung homogenates. mRNA data are presented as relative abundance compared with normal air-exposed WT controls. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. *P<0.05, **P<0.01; ***P<0.001; ****P<0.0001 compared with normal air-exposed WT or Tnfsf10−/− controls. #P<0.05; ##P<0.01; ###P<0.001; ####P<0.0001 compared with CS-exposed WT controls.
Active NF-κB p65 is reduced in TRAIL-deficient mice exposed to CS
We, and others, have previously shown that TRAIL induces inflammatory responses by mediating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activity.6, 28 In this study, CS exposure of WT mice increased the level of active NF-κB p65 in the lung compared with normal air-exposed WT controls (Figure 4i). In contrast and importantly, active NF-κB p65 did not increase in CS-exposed Tnfsf10−/− mice compared with normal air-exposed Tnfsf10−/− or WT controls. Consequently, active NF-κB p65 was decreased in CS-exposed Tnfsf10−/− compared with CS-exposed WT controls. We also assessed other NF-κB subunits (p50, p52 and RelB) but these were not altered by CS exposure or in the absence of TRAIL (data not shown).
Emphysema-like alveolar enlargement and cell death are reduced and lung function is improved in TRAIL-deficient mice exposed to CS
We previously showed that WT mice with experimental COPD have emphysema-like alveolar enlargement and impaired lung function.18, 19 Here, we again show that CS exposure of WT mice increased alveolar diameter compared with normal air-exposed WT controls (Figure 5a). CS exposure of Tnfsf10−/− mice also increased alveolar diameter compared with normal air-exposed Tnfsf10−/− controls. However, CS-exposed Tnfsf10−/− mice had significantly reduced alveolar diameter compared with CS-exposed WT controls. Reduced alveolar diameter in CS-exposed Tnfsf10−/− mice was associated with reduced numbers of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)+ cells in the parenchyma, indicating reduced apoptosis, compared with CS-exposed WT controls (Figure 5b).

Emphysema-like alveolar enlargement is reduced, apoptosis inhibited and lung function is preserved in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-deficient mice exposed to cigarette smoke (CS). Wild-type (WT) or TRAIL-deficient (Tnfsf10−/−) mice were exposed to CS or normal air for 8 weeks. (a) Alveolar diameter (μm) was determined in hematoxylin and eosin-stained lung sections using the mean linear intercept technique. (b) The numbers of TUNEL+ (terminal deoxynucleotidyl transferase dUTP nick end labeling) cells (arrowheads) enumerated in whole-lung sections. Lung function was assessed in terms of (c) pressure–volume loops and (d) lung compliance at 30 cmH2O. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. *P<0.05; **P<0.01; ****P<0.0001 compared with normal air-exposed WT or Tnfsf10−/− controls. ##P<0.01; ####P<0.0001 compared with CS-exposed WT controls.
We next assessed the role of TRAIL in impaired lung function. CS exposure of WT mice increased compliance, determined during a pressure–volume loop maneuver compared with normal air-exposed WT controls (Figure 5c,d). In contrast, CS exposure did not increase pressure–volume loops or lung compliance in Tnfsf10−/− mice compared with normal air-exposed Tnfsf10−/− controls or, importantly, CS-exposed WT controls.
Spontaneous small airway changes occur in TRAIL-deficient mice
We previously showed that mice develop small airway remodeling in experimental COPD.18, 19 Here, we replicate our previous observations and show that CS exposure of WT mice increased small airway epithelial cell area compared with normal air-exposed WT controls (Figure 6a). CS exposure of Tnfsf10−/− mice also increased small airway epithelial cell area compared with normal air-exposed Tnfsf10−/− controls, which was not different to CS-exposed WT smoke controls. Notably, however, small airway epithelial cell area increased spontaneously in normal air-exposed Tnfsf10−/− compared with WT controls.

Spontaneous airway remodeling occurs in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-deficient mice. Wild-type (WT) or TRAIL-deficient (Tnfsf10−/−) mice were exposed to cigarette smoke or normal air for eight weeks. (a) Small airway epithelial thickness in terms of epithelial cell area (μm2) per basement membrane (BM) perimeter (μm) was determined in periodic acid-Schiff (PAS)-stained whole-lung sections. (b) The number of epithelial cells in PAS-stained lung sections was assessed by enumerating the number of nuclei per 100 μm of BM perimeter. (c) Area of collagen deposition (μm2) per BM perimeter (μm) was determined in Masson’s Trichrome-stained lung sections. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. *P<0.05; ****P<0.0001 compared with normal air-exposed WT or Tnfsf10−/− controls. φφP<0.01; φφφP<0.001; φφφφP<0.0001 compared with normal air-exposed WT controls.
We then determined whether increased epithelial area was associated with increased numbers of nuclei in the small airways, which is an indicator of increases in the numbers of epithelial cells. Consistent with expanded small airway epithelial cell area, CS exposure of WT mice increased nuclei numbers in the small airways compared with normal air-exposed WT controls (Figure 6b). The numbers of nuclei in the small airways of CS-exposed Tnfsf10−/− mice were not different to normal air-exposed Tnfsf10−/− or CS-exposed WT controls. The former observation was because normal air-exposed Tnfsf10−/− controls had increased nuclei numbers in the small airways compared with WT air controls.
We next examined airway fibrosis in terms of collagen deposition around the small airways. CS exposure of WT mice increased collagen deposition compared with normal air-exposed WT controls (Figure 6c). CS exposure of Tnfsf10−/− mice did not alter collagen deposition compared with normal air-exposed Tnfsf10−/− or CS-exposed WT controls. The former observation was because normal air-exposed Tnfsf10−/− mice had increased collagen deposition compared with normal air-exposed WT air controls.
Pulmonary inflammation is suppressed and emphysema-like alveolar enlargement is inhibited in experimental COPD by therapeutic neutralization of TRAIL
We previously showed that the hallmark features of experimental COPD are emerging by week 6, established by week 8 and progressively worsen by week 12 of CS exposure.18 Hence, to assess the therapeutic potential of targeting TRAIL, WT mice were exposed to CS or normal air for 12 weeks and were treated with a neutralizing anti-TRAIL monoclonal antibody or isotype control intraperitoneally from week 7 to week 12.
We first examined the effect of TRAIL neutralization on pulmonary inflammation in BAL. As expected CS exposure of isotype-treated WT mice increased total leukocytes, macrophages, neutrophils, and lymphocytes in BAL compared with isotype-treated normal air-exposed WT controls (Figure 7a–d). CS exposure of anti-TRAIL-treated mice only partially increased total leukocytes, macrophages, neutrophils, and lymphocytes in BAL compared with anti-TRAIL-treated air controls. Furthermore, anti-TRAIL treatment significantly reduced all inflammatory cells compared with isotype-treated CS-exposed controls, although levels remained increased compared with normal air-exposed controls.

Pulmonary inflammation is suppressed and emphysema-like alveolar enlargement inhibited in experimental chronic obstructive pulmonary disease (COPD) by therapeutic neutralization of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Wild-type mice were exposed to cigarette smoke or normal air for twelve weeks and treated with neutralizing anti-TRAIL monoclonal antibody or isotype control, intraperitoneally three times per week, from week 7 to 12. (a) Total leukocytes, (b) macrophages, (c) neutrophils, and (d) lymphocytes were enumerated in May-Grunwald Giemsa-stained bronchoalveolar lavage (BAL). (e) The numbers of parenchymal inflammatory cells (arrowheads) were determined in periodic acid-Schiff-stained lung sections. (f) NF-κB p65 activity in whole-lung homogenates. (g) Alveolar diameter (μm) was determined in hematoxylin and eosin-stained lung sections using the mean linear intercept technique. (h) The numbers of TUNEL+ (terminal deoxynucleotidyl transferase dUTP nick end labeling) cells (arrowheads) enumerated in whole-lung sections. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. **P<0.01; ***P<0.001; ****P<0.0001 compared with isotype-treated or anti-TRAIL-treated normal air-exposed controls. #P<0.05; ##P<0.01; ###P<0.001; ####P<0.0001 compared with isotype-treated CS-exposed controls.
Next, we examined the effect of neutralizing TRAIL on inflammatory cell numbers in the parenchyma. As expected CS exposure of isotype-treated mice increased inflammatory cells in the parenchyma compared with isotype-treated normal air-exposed controls (Figure 7e). CS exposure of anti-TRAIL-treated mice partially increased inflammatory cells compared with anti-TRAIL-treated normal air-exposed controls. Furthermore, anti-TRAIL treatment significantly reduced inflammatory cells compared with isotype-treated CS-exposed controls.
Next, we assessed the effect of neutralizing TRAIL on active NF-κB p65 levels in the lung. Active NF-κB p65 was increased in CS-exposed isotype-treated mice compared with normal air-exposed isotype-treated controls (Figure 7f). In contrast, CS-exposed anti-TRAIL-treated mice completely inhibited the increase in active NF-κB p65 compared with anti-TRAIL-treated normal air-exposed or isotype-treated CS-exposed controls.
We then assessed the effect of neutralizing TRAIL on CS-induced emphysema-like alveolar enlargement. CS exposure of isotype-treated mice increased alveolar diameter compared with isotype-treated normal air-exposed controls (Figure 7g). In contrast, CS-exposed anti-TRAIL-treated mice were completely protected against increased alveolar diameter with no increase compared with anti-TRAIL-treated normal air-exposed or isotype-treated CS-exposed controls. Similarly, CS exposure of isotype-treated mice resulted in increased numbers of TUNEL+ cells in the parenchyma compared with normal air-exposed isotype-treated controls (Figure 7h). However, CS-exposed anti-TRAIL-treated mice were protected against increases in TUNEL+ cells in the parenchyma with no increase compared with CS-exposed isotype-treated controls.
Airway remodeling is suppressed in experimental COPD by therapeutic neutralization of TRAIL
We then assessed the effects of neutralizing TRAIL on airway remodeling in experimental COPD. As expected CS exposure of isotype-treated mice increased small airway epithelial cell area and nuclei numbers compared with isotype-treated normal air-exposed controls (Figure 8a,b). CS exposure of anti-TRAIL-treated mice only partially increased small airway epithelial cell area and nuclei numbers compared with anti-TRAIL-treated normal air-exposed controls. Furthermore, CS-exposed anti-TRAIL-treated mice had significantly reduced small airway epithelial cell area and nuclei numbers compared with isotype-treated CS-exposed controls. In addition, unlike in normal air-exposed Tnfsf10−/− mice, administration of anti-TRAIL to normal air-exposed WT mice did not increase airway epithelial cell area or nuclei numbers compared with isotype-treated controls.

Airway remodeling is reduced in experimental chronic obstructive pulmonary disease (COPD) by therapeutic neutralization of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Wild-type mice were exposed to cigarette smoke (CS) or normal air for twelve weeks and treated with neutralizing anti-TRAIL monoclonal or isotype control antibodies, intraperitoneally three times per week, from weeks 7 to 12. (a) Small airway epithelial thickness in terms of epithelial cell area (μm2) per basement membrane (BM) perimeter (μm) was determined in periodic acid-Schiff (PAS)-stained lung sections. (b) The number of epithelial cells in PAS-stained lung sections was assessed by enumerating the number of nuclei per 100 μm BM perimeter. (c) Area of collagen deposition (μm2) per BM perimeter (μm) in Masson’s Trichrome-stained lung sections. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. **P<0.01; ****P<0.0001 compared with anti-TRAIL or isotype normal air-exposed controls. #P<0.05; ###P<0.001 compared with isotype-treated CS-exposed controls.
Finally we examined the effect of neutralizing TRAIL on airway fibrosis. As expected CS exposure of isotype-treated mice increased collagen deposition around the small airways compared with isotype-treated normal air-exposed controls (Figure 8c). In contrast, CS-exposed anti-TRAIL-treated mice did not have increased collagen deposition compared with anti-TRAIL-treated normal air-exposed controls. In addition, unlike in normal air-exposed Tnfsf10−/− mice, administration of anti-TRAIL to normal air-exposed WT mice did not increase collagen deposition around the small airways compared with isotype-treated controls.
Discussion
In this study, we discovered a previously unrecognized role for TRAIL in CS-induced experimental COPD. TRAIL and its receptors were increased in mice with chronic CS-induced experimental COPD and in human COPD patients. Using a combination of CS exposure of WT and Tnfsf10−/− mice and a neutralizing antibody, we demonstrate that TRAIL increases pulmonary inflammation and expression of pro-inflammatory mediators, emphysema-like alveolar enlargement, and impairs lung function in experimental COPD. Inflammation and alveolar enlargement were associated with TRAIL-induced increases in active NF-κB p65 and apoptosis, respectively. Surprisingly, Tnfsf10−/− mice developed spontaneous airway remodeling characterized by increased epithelial area and collagen deposition. Importantly, therapeutic targeting of TRAIL with a neutralizing monoclonal antibody reduced CS-induced pulmonary inflammation and emphysema-like alveolar enlargement, without inducing airway remodeling (i.e., in normal air-exposed WT mice). This study advances the emerging knowledge of the roles of TRAIL in inflammatory and respiratory diseases, and its potential for therapeutic targeting.
To investigate the role of TRAIL in the pathogenesis of COPD, we used an established mouse model of experimental COPD.18, 19, 20, 21, 22, 23, 24 Mice were exposed via the nose only to tightly controlled doses of CS. This protocol induces the development of hallmark features of human COPD, namely chronic bronchitis (pulmonary inflammation), small airway remodeling, emphysema-like alveolar enlargement, and impaired lung function.22 Hence, our model recapitulates key pathological features observed in human disease.
We first showed that chronic CS exposure of WT mice resulted in concomitant increases in TRAIL mRNA and protein levels in the airways and parenchyma. Our examination of lung tissue sections stained for TRAIL identified airway epithelial cells and parenchymal-associated monocytes as sources of TRAIL. We have previously shown that mouse airway epithelial cells express TRAIL following allergen challenge or respiratory infection with Chlamydia murdiarum.6, 7, 17 Subsequent flow cytometric analysis of lung homogenates from mice with experimental COPD identified CD11b+ monocytes as a major source of cell surface-bound TRAIL. Consistent with these observations, we, and others, have shown that TRAIL-expressing macrophages are recruited to the lung following respiratory bacterial (e.g., Chlamydia, Streptococcus pneumoniae) and viral (e.g., influenza) infections.6, 8, 10 We also showed that TRAIL receptor expression was elevated in experimental COPD with increases in mRNA levels of DR5 in the airways and DcR1 in the parenchyma. Notably, in support of the data in mice there were also increases in the mRNA levels of TRAIL and its receptors DR4 and DR5 in human COPD lung tissue and airway epithelial cells.25, 26, 27 There were some species-specific differences in TRAIL receptor expression with increases in DcR1 in mice and DR4 in humans. Mice do not produce DR4, and so compensatory mechanisms may be present.
Tnfsf10−/− mice had reduced CS-induced pulmonary cellular inflammation characterized by reduced influx of total leukocytes and macrophages into the airways and decreased numbers of parenchymal-associated inflammatory cells and CD11b+ monocytes, mDCs, and γδT cells in the lung. All of these cells have been previously shown to be increased in experimental models of, or in human COPD.29, 30, 31, 32 These data are supported by our previous studies that show TRAIL drives pulmonary inflammation.6, 7, 16 Indeed, intranasal administration of recombinant TRAIL to naive mice increased the numbers of mDCs and CD4+ T cells in the lung.7 Furthermore, silencing of TRAIL using small interfering RNA reduced pulmonary inflammation in a mouse model of acute asthma (ovalbumin-induced AAD).7 We also showed that Tnfsf10−/− mice had reduced infiltration of CD11b+ monocytes, mDCs, CD4+, and CD8+ T cells into the lung following neonatal C. muridarum respiratory infection.6
In support of our inflammatory cell data, Tnfsf10−/− mice also had reduced CS-induced mRNA expression of key pro-inflammatory cytokines (TNF-α), chemokines (CCL2, 3, 7, 12, and 20) and other COPD-related factors (MMP-12 and SAA3) in the lungs. We, and others, have shown increased expression of TNF-α following CS exposure in mice and humans, and this cytokine is known to induce the expression of the monocyte chemokines CCL2, 7, and 12 and the neutrophil chemoattractant CCL3.18, 32, 33, 34 These chemokines have all been associated with increasing the severity of cellular inflammation and emphysema in COPD.35, 36 CCL20 was increased by CS and was decreased in Tnfsf10−/− mice, which correlated with decreased numbers of mDCs in the lung. We previously showed that inhibition of TRAIL reduced CCL20 and subsequent homing of mDCs to the airways, which was associated with reduced inflammation and airway hyperresponsiveness in AAD.7 We, and others, have also shown that the protease MMP-12 and SAA3, an acute phase protein, are increased in experimental and human COPD.18, 37, 38, 39 To investigate the mechanisms involved in TRAIL-mediated inflammation we assessed the activity of the transcription factor NF-κB. TRAIL is known to induce NF-κB activity and promote inflammatory responses.6, 17, 28 Consistent with these observations, our current study demonstrated that TRAIL deficiency or inhibition reduced CS-induced NF-κB p65 activity and this was associated with reduced pulmonary inflammation. This indicates that in CS-induced experimental COPD, TRAIL induces NF-κB p65 activity causing the transcription of the mRNA of pro-inflammatory factors that drive inflammation and disease pathogenesis. CS exposure has been shown to promote NF-κB nuclear localization in mice, which was associated with increased pulmonary inflammation and induction of multiple pro-inflammatory and COPD-related genes (e.g., TNF-α and MMP-12).34 We previously showed that TRAIL deficiency or administration of neutralizing anti-DcR2 antibody in mice reduced NF-κB activity and pulmonary inflammation in the lung following neonatal C. muridarum respiratory infection.6 We also recently showed that TRAIL induces the expression of the E3 ubiquitin ligase midline-1 in the airway wall and that siRNA-induced knockdown of midline-1 reduces NF-κB activity in AAD and RV infection models.17 Collectively, our current study advances our understanding of the roles of TRAIL as a pro-inflammatory mediator by showing that it regulates inflammation in CS-induced experimental COPD.
As in humans, chronic CS exposure causes emphysema-like alveolar enlargement in experimental COPD.18, 19, 20, 21, 22, 23, 24 Here we show that Tnfsf10−/− mice are protected against CS-induced alveolar enlargement. We also show that CS increases TRAIL+ monocytes and that Tnfsf10−/− mice have less TUNEL+ cells, indicating a reduction in apoptosis, in the parenchyma. Others have shown that TRAIL receptors are increased in the lungs of patients with emphysema and resected lung explant cultures from emphysematous patients or A549 cells exposed to CS extract displayed increased TRAIL-mediated apoptosis.40, 41 These data indicate that in CS-induced experimental COPD, TRAIL induces apoptosis of parenchymal cells that is involved in emphysema-like alveolar enlargement.
We then go on to show that the suppression of pulmonary inflammation and alveolar enlargement in the absence of TRAIL leads to protection against impaired lung function with the inhibition of increases in lung volumes and compliance. These are important features of human COPD. We previously showed that Tnfsf10−/− mice were protected against neonatal Chlamydia respiratory infection-induced emphysema-like alveolar enlargement and impaired lung function.6 TRAIL has also been shown to impair lung function in models of AAD.7, 16, 17
We previously showed that depletion of macrophages with clodronate-loaded liposomes reduced CS-induced airway remodeling, emphysema-like alveolar enlargement, and improved lung function in experimental COPD.18 Macrophages expressing TRAIL that are recruited to the lung during influenza virus infection induce acute lung injury and alveolar epithelial cell apoptosis.8 Collectively, these data indicate that TRAIL induced pulmonary inflammation and that TRAIL+ monocytes may contribute to CS-induced emphysema by promoting apoptosis in the parenchyma, which together reduce lung function.
Surprisingly, naive Tnfsf10−/− mice had spontaneous small airway changes characterized by increased airway epithelial cell area and numbers and collagen deposition, which were not further increased by CS exposure. In this regard, we previously showed that TRAIL induced airway epithelial thickening and goblet cell metaplasia in neonatal Chlamydia respiratory infection and AAD in mice.6 Others have shown that Tnfsf10−/− mice had increased total collagen in the lung in a bleomycin-induced mouse model of pulmonary fibrosis.9 Furthermore, recombinant TRAIL treatment inhibited the expression of the collagen-specific molecular chaperone heat-shock protein 47 and reduced soluble collagen production in human hepatic stellate cells in vitro.42 In contrast to these findings, however, Tnfsf10−/− mice had reduced lung collagen in a chronic ovalbumin model of AAD.16 These differences may result from the use of female versus male mice, and focused on the small rather than central airways in the current compared with the previous16 study.
Importantly, anti-TRAIL treatment of established experimental COPD substantially reduced CS-induced pulmonary inflammation, alveolar enlargement, and small airway remodeling. Anti-TRAIL treatment of normal air-exposed WT mice did not induce airway remodeling. This is in contrast to the small airway changes that occurred in naive Tnfsf10−/− mice. This suggests that therapeutic neutralization of TRAIL may be effective in suppressing hallmark features of COPD, without causing unwanted airway remodeling that occurs with early life or lifelong deficiency. Treatment may be further improved with increasing doses or optimizing treatment regimes. The therapeutic benefit in suppressing TRAIL is supported by our previous studies. We showed that TRAIL neutralization protected mice from neonatal Chlamydia respiratory infection-induced pulmonary inflammation and emphysema-like alveolar enlargement.6 Moreover, inhibition of TRAIL suppressed inflammation and improved lung function in mouse models of AAD.7, 17 Long-term selective inhibition of TRAIL may be contraindicated as a treatment for COPD due to the increased risk of lung cancer and respiratory infections.10 However, these issues may be overcome by optimizing dosing regimes. TRAIL is an important inducer of apoptosis in a variety tumor cells.43 Furthermore, both COPD patients and mice with experimental COPD are more susceptible to bacterial (e.g., Streptococcus pneumoniae) and viral (e.g., influenza) infections18, 20, 44 and TRAIL is required for the clearance of such infections.10, 45 These potential side effects could be minimized by short-term dosing or with optimized treatment regimes. Furthermore, additional studies may identify specific signaling factors and pathways downstream of TRAIL, which when inhibited do not increase cancer risk or susceptibility to infection. This could be initially assessed in mouse models of cancer and infections.
In summary, our study reveals for the first time that TRAIL has important pathogenic roles in CS-induced experimental COPD. It is pivotal in promoting pulmonary inflammation and NF-κB p65 activation, and emphysema-like alveolar enlargement and apoptosis, which lead to impaired lung function (Supplementary Figure S6). TRAIL also regulates small airway remodeling independent of CS exposure. Importantly, therapeutic neutralization of TRAIL in established experimental COPD reduced hallmark features of the disease. Collectively, our data suggest that therapeutic targeting of TRAIL may be beneficial in COPD.
Methods
Experimental COPD. Female, 7–8-week old, WT or Tnfsf10−/− BALB/c mice were exposed to normal air or CS through the nose only for 8 weeks as we have previously described.18, 19, 20, 21, 22, 23 Some mice were exposed to CS or normal air for 12 weeks and treated with anti-TRAIL neutralizing or isotype control antibodies from week 7 to 12.18 In recent years some studies have shown that COPD prevalence and mortality is higher in females, and in the United States in 2009 women accounted for 53% of COPD deaths. It is for this and logistics reasons that female mice are used.46
Isolation of RNA and protein. Total RNA was extracted from whole-lung tissue and blunt-dissected airway and parenchyma as described previously.17 Briefly, the trachea and lungs were excised, and lung parenchyma was carefully separated from the airways with sterile forceps. Whole lungs, airways, and parenchyma were then snap frozen and stored at −80 °C. Tissues were thawed and homogenized in 500 μl of sterile Dulbecco’s phosphate-buffered saline (Life Technologies, Mulgrave, VIC, Australia) using a Tissue-Tearor stick homogenizer (BioSpec Products, Bartesville, OK) on ice. Tissue homogenates were then split equally (250 μl) for RNA and protein extraction. Total RNA was extracted using TRIzol (Invitrogen, Mount Waverly, VIC, Australia) according to manufacturer’s instructions and stored at −80 °C. For protein extraction, tissue homogenates were mixed with equal portions of sterile Dulbecco’s phosphate-buffered saline (Life Technologies) supplemented with PhosSTOP phosphatase and Complete ULTRA protease inhibitors cocktails (Roche Diagnostics, Mannheim, Germany). Tissue homogenates were then centrifuged at 8,000 g for 10 min at 4 °C. Supernatants were collected and stored at −20 °C for assessment by ELISA assay.
qPCR. Total RNA from whole lungs, airway, and parenchyma (1,000 ng) were reversed transcribed using Bioscript (Bioline, Alexandria, NSW, Australia) and random hexamer primers (Invitrogen).6, 19, 47, 48, 49, 50 The mRNA expression of TRAIL, DR5, DcR1, TNF-α, CCL2, 3, 7, 12 and 20, MMP-12, SAA3, DcR2, IL-33, CXCL1, CXCL3, CCL4, CCL22, and mucin 5ac were determined by real-time quantitative PCR (qPCR, ABIPrism7000, Applied Biosystems, Scoresby, VIC, Australia) and expressed as relative abundance to the reference gene hypoxanthine-guanine phosphoribosyltransferase.6, 19, 47, 48, 49, 50 Custom designed primers were used (Supplementary Table 1).
ELISA. TRAIL protein levels in airway and parenchyma were quantified with mouse TRAIL/TNFSF10 DuoSet ELISA kits (R&D Systems, Gymea, NSW, Australia) with normalization to total protein determined using the BCA Protein Assay Kit (PIERCE, Scorsby, VIC, Australia) as per manufacturer’s instructions.
NF-κB assays. The active NF-κB p65 subunit was measured using the TransAM NF-κB family transcription factor assay kit (Active Motif, Carlsbad, CA) according to the manufacturer’s instructions.6, 17
Immunohistochemistry. Lungs were perfused, inflated, formalin-fixed, paraffin-embedded, and sectioned (4–6μm). Longitudinal sections of the left lung were incubated with primary antibody (anti-TRAIL, Abcam, Melbourne, VIC, Australia) overnight at 4 °C and followed by anti-rabbit horseradish peroxidase-conjugated secondary antibody (R&D Systems) as per manufacturer’s instructions. 3,3′-Diaminobenzidine chromogen-substrate buffer (DAKO, North Sydney, NSW, Australia) was applied to sections and incubated. Sections were counterstained with hematoxylin, mounted and analyzed with a BX51 microscope (Olympus, Tokyo, Shinjuku, Japan) and Image-Pro Plus software (Media Cybernetics, Rockville, MD).
Flow cytometry. Numbers of CD11b+ monocytes, neutrophils, γδT cells, natural killer T cells, CD4+ T cells, CD8+ T cells, mDCs, alveolar macrophages, and pDCs, in lung homogenates were determined based on surface marker expression using flow cytometry (Table 1).6, 48, 51, 52 Flow cytometric analysis was performed using a FACSAriaIII with FACSDiva software (BD Biosciences, North Ryde, NSW, Australia). Flow cytometry antibodies were from Biolegend (Karrinyup, WA, Australia) (Supplementary Table 2). OneComp compensation beads (eBioscience) were used to set up assays.
Analysis of differential gene expression. Differential gene expression analysis of published datasets (accession numbers GSE5058 and GSE27597)25, 26, 27 was performed with the Array Studio software (Omicsoft Corporation, Research Triangle Park, NC) applying a general linear model adjusting for age and gender and the Benjamini–Hochberg method for P value adjustment.
Airway and parenchymal inflammation. Airway inflammation was assessed by differential enumeration of inflammatory cells in BAL.18, 50, 51, 53, 54 Longitudinal sections of lung were stained with periodic acid-Schiff and parenchymal inflammation was assessed by enumerating the numbers of inflammatory cells in 20 randomized, high-powered fields.18
Alveolar enlargement. Lungs were perfused, inflated, fixed, paraffin embedded, and sectioned (4–6μm). Longitudinal sections of the left single-lobe lung were stained with hematoxylin and eosin to assess alveolar diameter using the mean linear intercept technique.6, 18, 19, 20, 21, 49
TUNEL assay. Longitudinal sections of the left single-lobe lung were stained with TUNEL assay kits (Promega, Sydney, NSW, Australia) according to manufacturer’s instructions. Apoptosis in lung parenchyma was assessed by enumerating the numbers of TUNEL+ cells in 20 randomized, high-powered fields.
Lung function. Lung compliance was assessed by quasi-static pressure–volume loops from oscillation maneuvers (Flexivent (SCIREQ, Montreal, QC, Canada)) as the volume of air that entered the lungs when the airway pressure was increased from 2 to 30 cmH2O by the ventilator (PVs-P Flexivent maneuver). Compliance was calculated as the measured change in volume divided by this applied pressure change. Three inflations were performed and averaged per mouse.55
Airway remodeling. Longitudinal sections of the left single-lobe lung were stained with periodic acid-Schiff or Masson’s Trichrome. Airway epithelial area (μm2) and cell (nuclei) number, and collagen deposition area (μm2) was assessed in a minimum of four small airways (basement membrane perimeter <1,000 μm) per section.16, 18, 19 Data were normalized to basement membrane perimeter (μm) and quantified using ImageJ software (Version 1.49h, NIH, New York City, NY).
TRAIL neutralization. Mice were treated with 12.5 mg kg−1 body weight of neutralizing anti-TRAIL monoclonal antibody (clone N2B2) or rat IgG2a isotype control (clone 2A3, BioXCell, West Lebanon, NH) from week 7 to 12 (six weeks), by intraperitoneally injections three times per week.6
Statistical analysis. Data (n=5–6) presented as means±s.e.m. are representative of two independent experiments. Statistical significance was determined with two-tailed Mann–Whitney test or by one-way analysis of variance with Bonferroni post-test using GraphPad Prism Software version 6 (San Diego, CA).
Ethics statement. This study was performed in accordance with the recommendations issued by the National Health and Medical Research Council of Australia. All protocols were approved by the animal ethics committee of The University of Newcastle, Australia.
References
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012).
Han, M.K. et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am. J. Respir. Crit. Care Med. 182, 598–604 (2010).
Keely, S., Talley, N.J. & Hansbro, P.M. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol. 5, 7–18 (2012).
Willemse, B., Postma, D., Timens, W. & ten Hacken, N. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur. Respir. J. 23, 464–476 (2004).
Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 131, 636–645 (2013).
Starkey, M. et al. Tumor necrosis factor-related apoptosis-inducing ligand translates neonatal respiratory infection into chronic lung disease. Mucosal Immunol. 7, 478–488 (2013).
Weckmann, M. et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat. Med. 13, 1308–1315 (2007).
Herold, S. et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 205, 3065–3077 (2008).
McGrath, E.E. et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax 67, 796–803 (2012).
Steinwede, K. et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J. Exp. Med. 209, 1937–1952 (2012).
Stary, G. et al. Plasmacytoid dendritic cells express TRAIL and induce CD4+ T-cell apoptosis in HIV-1 viremic patients. Blood 114, 3854–3863 (2009).
Pan, G. et al. The receptor for the cytotoxic ligand TRAIL. Science 276, 111–113 (1997).
Sheridan, J.P. et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277, 818–821 (1997).
Pan, G. et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 (1997).
Collison, A., Foster, P.S. & Mattes, J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clin. Exp. Pharmacol. Physiol. 36, 1049–1053 (2009).
Collison, A. et al. Tumor necrosis factor-related apoptosis-inducing ligand regulates hallmark features of airways remodeling in allergic airways disease. Am. J. Respir. Cell Mol. Biol. 51, 86–93 (2014).
Collison, A. et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat. Med. 19, 232–237 (2013).
Beckett, E.L. et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 131, 752–762 (2013).
Hansbro, P.M. et al. Importance of mast cell Prss31/transmembrane tryptase/tryptase-γ in lung function and experimental chronic obstructive pulmonary disease and colitis. J. Biol. Chem. 289, 18214–18227 (2014).
Hsu, A. C.-Y. et al. Targeting PI3K-p110α suppresses influenza viral infection in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 191, 1012–1023 (2015).
Franklin, B.S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737 (2014).
Fricker, M., Deane, A. & Hansbro, P.M. Animal models of chronic obstructive pulmonary disease. Expert Opin. Drug Discov. 9, 629–645 (2014).
Tay, H.L. et al. Antagonism of miR-328 increases the antimicrobial function of macrophages and neutrophils and rapid clearance of non-typeable Haemophilus influenzae (NTHi) from infected lung. PLoS Pathog. 11, e1004549 (2015).
Vlahos, R. & Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. (Lond) 126, 253–265 (2014).
Campbell, J.D. et al. A gene expression signature of emphysema-related lung destruction and its reversal by the tripeptide GHK. Genome Med. 4, 67 (2012).
Christenson, S.A. et al. miR-638 regulates gene expression networks associated with emphysematous lung destruction. Genome Med. 5, 114 (2013).
Tilley, A.E. et al. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 179, 457–466 (2009).
Degli-Esposti, M.A. et al. The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7, 813–820 (1997).
Botelho, F.M. et al. Cigarette smoke-induced accumulation of lung dendritic cells is interleukin-1α-dependent in mice. Respir. Res. 13, 81 (2012).
Shan, M. et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 4, 117ra9 (2012).
Duan, M. et al. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol. 189, 946–955 (2012).
Bozinovski, S. et al. Innate cellular sources of interleukin-17 A regulate macrophage accumulation in cigarette- smoke-induced lung inflammation in mice. Clin. Sci. (Lond) 129, 785–796 (2015).
Vlahos, R. et al. Neutralizing granulocyte/macrophage colony-stimulating factor inhibits cigarette smoke-induced lung inflammation. Am. J. Respir. Crit. Care Med. 182, 34–40 (2010).
Vlahos, R. et al. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L931–L945 (2006).
Frankenberger, M. et al. Chemokine expression by small sputum macrophages in COPD. Mol. Med. 17, 762–770 (2011).
Chen, K. et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One 6, e20333 (2011).
Bozinovski, S. et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 109, 935–940 (2012).
Demedts, I.K. et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax 61, 196–201 (2006).
Anthony, D. et al. Serum amyloid A promotes lung neutrophilia by increasing IL-17A levels in the mucosa and γδ T cells. Am. J. Respir. Crit. Care Med. 188, 179–186 (2013).
Morissette, M.C., Parent, J. & Milot, J. The emphysematous lung is abnormally sensitive to TRAIL-mediated apoptosis. Respir. Res. 12, 105 (2011).
Morissette, M.C., Vachon-Beaudoin, G., Parent, J., Chakir, J. & Milot, J. Increased p53 level, Bax/Bcl-x(L) ratio, and TRAIL receptor expression in human emphysema. Am. J. Respir. Crit. Care Med. 178, 240–247 (2008).
Park, S.-J., Sohn, H.-Y. & Park, S.I. TRAIL regulates collagen production through HSF1-dependent Hsp47 expression in activated hepatic stellate cells. Cell Signal. 25, 1635–1643 (2013).
Yagita, H., Takeda, K., Hayakawa, Y., Smyth, M.J. & Okumura, K. TRAIL and its receptors as targets for cancer therapy. Cancer Sci. 95, 777–783 (2004).
Sethi, S. et al. Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 176, 356–361 (2007).
Brincks, E.L., Katewa, A., Kucaba, T.A., Griffith, T.S. & Legge, K.L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 181, 4918–4925 (2008).
Pinkerton, K.E. et al. Women and lung disease. Sex differences and global health disparities. Am. J. Respir. Crit. Care Med. 192, 11–16 (2015).
Horvat, J.C. et al. Early-life chlamydial lung infection enhances allergic airways disease through age-dependent differences in immunopathology. J. Allergy Clin. Immunol. 125, 617–625 (2010).
Starkey, M.R. et al. Constitutive production of IL-13 promotes early-life Chlamydia respiratory infection and allergic airway disease. Mucosal Immunol. 6, 569–579 (2013).
Beckett, E.L. et al. TLR2, but not TLR4, is required for effective host defence against Chlamydia respiratory tract infection in early life. PLoS One 7, e39460 (2012).
Essilfie, A.-T. et al. Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax 70, 458–467 (2015).
Horvat, J.C. et al. Chlamydial respiratory infection during allergen sensitization drives neutrophilic allergic airways disease. J. Immunol. 184, 4159–4169 (2010).
Thorburn, A.N., Foster, P.S., Gibson, P.G. & Hansbro, P.M. Components of Streptococcus pneumoniae suppress allergic airways disease and NKT cells by inducing regulatory T cells. J. Immunol. 188, 4611–4620 (2012).
Essilfie, A.-T. et al. Haemophilus influenzae infection drives IL-17-mediated neutrophilic allergic airways disease. PLoS Pathog. 7, e1002244 (2011).
Asquith, K.L. et al. Interleukin-13 promotes susceptibility to chlamydial infection of the respiratory and genital tracts. PLoS Pathog. 7, e1001339 (2011).
Harris, R.S. Pressure-volume curves of the respiratory system. Respir. Care 50, 78–98 (2005).
Acknowledgements
This work was supported by grants and fellowships from the National Health and Medical Research Council of Australia, Rebecca L. Cooper Medical Research Foundation and a Gladys Brawn Fellowship. TRAIL-deficient mice were obtained from Amgen (Amgen, Seattle, WA). APC-conjugated αGalCer tetramers were obtained from Professor Dale Godfrey, University of Melbourne, Australia. We thank Kristy Wheeldon for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article

Cite this article
Haw, T., Starkey, M., Nair, P. et al. A pathogenic role for tumor necrosis factor-related apoptosis-inducing ligand in chronic obstructive pulmonary disease. Mucosal Immunol 9, 859–872 (2016). https://doi.org/10.1038/mi.2015.111
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.111
This article is cited by
-
Airway and parenchyma transcriptomics in a house dust mite model of experimental asthma
Respiratory Research (2023)
-
TRAIL protects the immature lung from hyperoxic injury
Cell Death & Disease (2022)
-
A monoclonal antibody to Siglec-8 suppresses non-allergic airway inflammation and inhibits IgE-independent mast cell activation
Mucosal Immunology (2021)
-
TRAIL signals through the ubiquitin ligase MID1 to promote pulmonary fibrosis
BMC Pulmonary Medicine (2019)
-
Microbiome effects on immunity, health and disease in the lung
Clinical & Translational Immunology (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}