
Abstract
Chronic gastritis from Helicobacter pylori infection is a major factor in the development of gastric adenocarcinoma. Factors that regulate gastritis severity are important in determining which individuals are susceptible to H. pylori–associated disease. Although protease-activated receptor 1 (PAR1) has been identified as one such host factor, its mechanism of action is unknown. Using chimeric mice, we demonstrated that PAR1-mediated protection against H. pylori gastritis requires bone marrow–derived cells. Analyses of the gastric mucosa revealed that PAR1 suppresses cellular infiltration and both T helper type 1 (Th1) and T helper type 17 (Th17) responses to infection. Moreover, PAR1 expression was associated with reduced vaccine-mediated protection against H. pylori. Analyses of H. pylori–stimulated macrophages revealed that PAR1 activation suppressed secretion of interleukin (IL)-12 and IL-23, key drivers of Th1 and Th17 immunity, respectively. Furthermore, PAR1 suppressed interferon regulatory factor 5 (IRF5), an important transcription factor for IL-12 and IL-23, both in the infected mucosa and following bacterial stimulation. PAR1 suppression of IRF5 and IL-12/23 secretion by macrophages provides a novel mechanism by which the host suppresses the mucosal Th1 and Th17 response to H. pylori infection. Dysregulation of this process is likely an important factor in the susceptibility of some individuals to H. pylori–associated disease.
Similar content being viewed by others
INTRODUCTION
The pathological consequences of infection by Helicobacter pylori, including gastric adenocarcinoma and peptic ulcer disease,1, 2 occur as a result of the chronic inflammation that develops from long-term colonization by these pathogenic bacteria. This gastritis is characterized by a mixed T helper type 1 (Th1) and T helper type 17 (Th17)-type immune response, and requires the presence of macrophages.3, 4, 5, 6, 7 Not all H. pylori–infected individuals develop disease, and factors that modify the severity of resulting inflammation appear to play a key role in determining an individual’s disease susceptibility. Progression to disease is highly complex and is influenced by an interaction of environmental factors, variability in H. pylori virulence factors including the cag pathogenicity island,8 as well as host genetic factors9, 10 such as cytokine polymorphisms.11, 12
One such host factor is the protease-activated receptor 1 (PAR1). PARs comprise a family of cell surface G protein–coupled receptors that are expressed on a wide range of cell types. PAR activation occurs via a unique mechanism of activation involving cleavage of an extracellular domain by a specific serine protease, unmasking a tethered ligand that binds to the body of the PAR.13 This initiates a signaling cascade that can result in a variety of cell responses, including proliferation, apoptosis, and/or the production of cytokines and prostanoids.14, 15, 16 PAR-activating proteases are derived from numerous sources including epithelial and inflammatory cells, coagulation factors, and bacteria. Activation of PAR by serine proteases, released during infection and inflammation, have been shown to play essential roles in regulating the innate immune response to infection.16
We have previously shown that PAR1 is a critical suppressor of H. pylori–induced gastritis in an animal model, with infected PAR1-deficient mice developing a severe atrophic gastritis, in contrast to controls expressing PAR1 that only developed mild gastritis.17 The clinical relevance of this observation is supported by studies from us and others that have associated human PAR1 polymorphisms with susceptibility to gastric cancer.18, 19 However, as PAR1 is expressed by numerous cell types, including epithelial, endothelial, smooth muscle, and immune cells, the source and precise mechanism of its protective activity is unknown.
Here we identify this mechanism by showing that PAR1 inhibits both Th1 and Th17 immune responses to H. pylori in the gastric mucosa, and that suppression of these responses is mediated by PAR1 activation inhibiting the secretion of proinflammatory cytokines by macrophages. Furthermore, we demonstrate that although PAR1 is protective against the pathogenic effects of H. pylori, it actually inhibits the ability of vaccinations to combat this infection.
RESULTS
H. pylori gastritis is regulated by PAR1-expressing bone marrow–derived cells
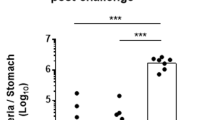
Previously, we showed that at 2 months after infection, PAR1−/− mice are colonized by significantly less H. pylori than wild-type controls and develop more severe pathology,17 although the cells responsible for this effect were not identified. PAR1 is expressed by a range of cell types found in the stomach, including gastric epithelial cells and a variety of immune cells.15, 16 Using bone marrow chimeras, we now show that mice reconstituted with PAR1−/− bone marrow are colonized by significantly fewer H. pylori than mice receiving wild-type bone marrow (Figure 1a). No significant increase in immune cell infiltration was detected in the infected gastric mucosa of mice receiving PAR1−/− bone marrow (Figure 1b). However, mice reconstituted with PAR1−/− bone marrow developed severe atrophic gastritis upon infection (median atrophy scores of 3) in stark contrast to mice receiving wild-type bone marrow that only typically developed mild atrophy (median atrophy score of 1; Figure 1b–e). Notably, only mice reconstituted with PAR1−/− bone marrow developed severe atrophic gastritis (Figure 1c–f) and this was irrespective of the PAR1 status of the recipient, indicating PAR1 regulation of H. pylori–induced gastritis was mediated by bone marrow–derived immune cell populations.

Helicobacter pylori–induced atrophic gastritis is associated with protease-activated receptor 1 (PAR1) expression on bone marrow–derived cells. Irradiated wild-type (WT) or PAR1−/− mice (n=5–7) were reconstituted with WT or PAR1−/− bone marrow for 2 months before infection with H. pylori. (a) At 2 months after infection, stomachs were removed and H. pylori colonization determined by colony-forming assay. Mice reconstituted with PAR1−/− bone marrow had significantly less H. pylori colonization than WT controls reconstituted with WT bone marrow (*analysis of variance (ANOVA)). (b–d) Severity of inflammation was scored histologically for cell infiltrate (0–6) and atrophy (0–3). (b, c) Cell infiltrate and atrophy scores for separate groups are shown, whereas (d) presents scores with all mice grouped according to the genotype of their bone marrow donor. Mice reconstituted with PAR1−/− bone marrow developed more severe atrophic gastritis than those receiving WT bone marrow (*Mann–Whitney). Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars). Representative pictures of histology are shown of typical infected mice transplanted with (e) WT bone marrow and (f) PAR1−/− bone marrow. Bars=200 μm.
PAR1 suppresses the T helper response to H. pylori infection in vivo
As a key feature of H. pylori–induced gastritis is the induction of a mixed Th1 (i.e., interferon-γ (IFNγ)) and Th17 (i.e., interleukin (IL)-17A) response,3, 4, 5, 6 we examined the effect of PAR1 on production of these cytokines in the infected mucosa. Gastritis in PAR1−/− mice does not develop until after 1 month of infection with H. pylori.17 Gastric tissues from mice with severe gastritis would be expected to contain increased levels of proinflammatory cytokines compared with mice with mild gastritis. In order to measure the direct regulatory effects of PAR1 on the host response to infection, without the complication of cytokine levels increasing because of gastritis, we compared cytokine levels in the stomachs of wild-type and PAR1−/− mice at 1 week after infection.
One week after infection, both IFNγ and IL-17A were significantly elevated in PAR1−/− mice relative to wild-type controls (Figure 2a,b). The increased cytokine levels in infected PAR1−/− mice were not because of a failure of these mice to produce local IL-10 (Figure 2c), which is known to be an important regulatory cytokine in this infection. Analysis of the serum antibody response revealed that PAR1−/− mice infected with H. pylori for 2 months had a significantly higher IgG2c but not IgG1 response than wild-type mice (Figure 2d), supportive of an increased Th1-type immune response in PAR1−/− animals.

Protease-activated receptor 1 (PAR1) suppression of T helper cytokines in gastric tissues from mice with acute Helicobacter pylori infection. (a–c) Cytokine levels in the gastric mucosa of wild-type (WT) or PAR1−/− mice, either uninfected (n=3) or infected (n=4) with H. pylori for 1 week, were quantified by enzyme-linked immunosorbent assay (ELISA). (d) Sera collected from WT and PAR1−/− mice infected with H. pylori for 2 months (n=8) were analyzed by ELISA for immunoglobulin G (IgG) subclass antibody response. Uninfected control levels were subtracted. Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars). H. pylori–infected PAR1−/− mice had significantly higher levels of gastric interferon-γ (IFNγ), interleukin (IL)-17A, and serum IgG2c than infected WT controls (*analysis of variance (ANOVA)). IL-10 levels were not significantly different (n.s.).
Flow cytometric analysis was used to quantify immune cells infiltrating into the gastric tissues of PAR1−/− mice infected with H. pylori for 1 week. This revealed a small but significant increase in the infiltration of CD4+ T cells, B cells, neutrophils, and macrophages into the PAR1−/− stomach, relative to wild-type mice (Figure 3). The stomachs of infected PAR1−/− mice had significantly increased levels of CD4+ T cells, B cells, and neutrophils compared with uninfected controls. There was a trend toward increased macrophage numbers, but this did not reach significance in this study (Figure 3).

Immune cell infiltration in Helicobacter pylori–infected PAR1−/− mice. Gastric cells perfused from the stomachs of uninfected (n=4) and 1-week H. pylori–infected (n=5/6) wild-type and PAR1−/− mice were analyzed by flow cytometry. (a) Identification of immune cell subsets was as illustrated: T helper (Th) cells were CD45+CD4+CD19−, B cells were CD45+CD4−CD19+, neutrophils were CD45+Ly6G+CD11b+, and macrophages were CD45+Ly6G−CD11b+MHC-II+,CD19− (filled histogram=anti-CD19; empty histogram=control cells without anti-CD19 antibody). (b) Increased cell infiltration in infected PAR1−/− mice (analysis of variance (ANOVA)). PAR1, protease-activated receptor 1.
In a further extension of this analysis and in order to evaluate the mucosal immune response to this infection draining the gastrointestinal tract, we also quantified cytokine levels in mesenteric lymph nodes from uninfected and infected mice. Once again, both IFNγ and IL-17A were significantly elevated in PAR1−/− mice relative to wild-type controls (Figure 4), whereas no significant increase was observed in the Th2 cytokine IL-13, the immunosuppressive cytokines IL-10 and transforming growth factor-β, or in proinflammatory macrophage inflammatory protein-2 (MIP-2). The absence of an increased Th2 response in PAR1−/− mice is consistent with the similarity of the immunoglobulin G1 (IgG1) antibody response between these animals and wild-type mice.

Increased T helper type 1 (Th1) and T helper type 17 (Th17) cytokines in lymphoid tissue draining the mucosal gastrointestinal tract of Helicobacter pylori–infected PAR1−/− mice. Cytokine levels in mesenteric lymph nodes (MLNs) of wild-type (WT) or PAR1−/− mice, either uninfected (n=4) or infected (n=8) with H. pylori for 1 month, were quantified by enzyme-linked immunosorbent assay (ELISA). Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars). H. pylori–infected PAR1−/− mice had significantly higher levels of lymphoid interferon-γ (IFNγ) and interleukin (IL)-17A than infected WT controls (*analysis of variance (ANOVA)), but not other cytokines tested. PAR1, protease-activated receptor 1.
Vaccination-mediated protection against H. pylori is enhanced in the absence of PAR1
IL-17 is potentially a key cytokine in vaccine-mediated protection against H. pylori.20, 21 As our data indicated that PAR1 regulated the IL-17 response to H. pylori in vivo (Figure 2b), this raised the possibility that PAR1 activity may negatively affect vaccine effectiveness against H. pylori. To test this possibility, PAR1−/− and wild-type mice were orally vaccinated before challenge with live bacteria. Consistent with the antibody response to infection (Figure 2d), vaccinated and challenged PAR1−/− mice mounted a significantly higher H. pylori–specific serum IgG2c but not IgG1 response than wild-type controls (Figure 5a). In this study we also evaluated the intestinal antibody response and found IgG2c was also the only antibody class significantly increased in intestinal scrapings from vaccinated and challenged PAR1−/− mice (Figure 5b).

Increased efficacy of vaccinations against Helicobacter pylori in PAR1−/− mice. Wild-type (WT) or PAR1−/− mice (n=8) were orally vaccinated against H. pylori. Unvaccinated controls were sham-treated with phosphate-buffered saline (PBS). At 4 weeks after vaccination, mice were challenged with live H. pylori and samples collected a further 4 weeks later. Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars). (a) Serum and (b) intestinal anti-H. pylori antibody responses were quantified by enzyme-linked immunosorbent assay (ELISA). Vaccinated/challenged PAR1−/− mice had a significantly higher serum and intestinal IgG2c response than WT controls (*analysis of variance (ANOVA)). (c) Efficacy of vaccination was determined by colony-forming assay. Vaccination of both WT and PAR1−/− mice resulted in a significant reduction in H. pylori colonization as compared with unvaccinated controls (*ANOVA). The percentage of protective immunity induced by vaccination of individual WT and PAR1−/− mice was calculated using the following formula: (100−(CFU of vaccinated mouse/median of unvaccinated control group)) × 100. The percentage reduction of H. pylori colonization in vaccinated PAR1−/− mice was significantly higher than that resulting from vaccination of WT mice (*Mann–Whitney). CFU, colony-forming unit; PAR1, protease-activated receptor 1.
Vaccination significantly reduced H. pylori numbers in both wild-type and PAR1−/− mice compared with sham-vaccinated controls (Figure 5c). However, vaccination of PAR1−/− mice resulted in an average 93% reduction in H. pylori colonization compared with only a 63% reduction in wild-type mice (Figure 5d). Hence, vaccination was significantly more protective against H. pylori colonization in mice lacking PAR1, supporting our hypothesis that PAR1-mediated suppression of cytokines opposes vaccine efficacy.
PAR1 inhibits Th1 and Th17 cytokine secretion by suppressing macrophage production of IL-12 and IL-23
To determine whether concurrent PAR1 activation was required for Th1 and Th17 inhibition, we evaluated the in vitro cytokine response of murine splenocytes stimulated with H. pylori. Addition of a PAR1-activating but not control peptide significantly suppressed the IFNγ and IL-17A response of splenocytes stimulated with H. pylori for 24 h (Figure 6a). However, this effect was preceded by PAR1-mediated suppression of both IL-12 (a key inducer of IFNγ) and IL-23 (a key inducer of IL-17) at 18 h after H. pylori stimulation (Figure 6b).

Protease-activated receptor 1 (PAR1)–mediated suppression of T helper type 1 (Th1) and T helper type 17 (Th17) cytokine production is preceded by inhibition of interleukin (IL)-12 and IL-23 secretion. Splenocytes from wild-type mice (n=7) were stimulated with Helicobacter pylori SS1. (a, b) After 18 and 24 h, cytokines secreted into supernatants were quantified by enzyme-linked immunosorbent assay (ELISA), with background levels from unstimulated cultures subtracted. Addition of a PAR1-activating but not a control scrambled peptide significantly reduced cytokine secretion (*analysis of variance (ANOVA)). Data presented are from one of three similar experiments. Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars).
These data indicated that PAR1 activation might not directly suppress T cells, but rather act upstream by inhibiting the secretion of IL-12 and IL-23. The main cell types that secrete IL-12 and IL-23 are macrophages and dendritic cells (DCs). Despite repeated attempts (six experiments) we found no evidence that PAR1 activation inhibits the secretion of IL-12 or IL-23 by DCs (Figure 7a). However, PAR1 activation reproducibly suppressed cytokine secretion by macrophages (Figure 7b). Importantly, we showed that H. pylori–induced secretion of IFNγ (a cytokine produced by T cells and innate immune cells, i.e., natural killer cells) by splenocytes was macrophage dependent, as depletion of macrophages blocked release of this cytokine (Figure 7c). Moreover, the suppressive effects of PAR1 peptide on IFNγ secretion could be reversed by the addition of recombinant IL-12 (Figure 7c).

Protease-activated receptor 1 (PAR1) activation suppresses macrophage secretion of interleukin (IL)-12 and IL-23. (a) Bone marrow–derived dendritic cells (DCs) and (b) peritoneal macrophages from wild-type (WT) mice (n=4) were stimulated with Helicobacter pylori SS1 for 12 or 18 h, and then secreted cytokines quantified by enzyme-linked immunosorbent assay (ELISA). Addition of a PAR1-activating (TF) but not control scrambled (FT) peptide significantly reduced cytokine secretion by macrophages (analysis of variance (ANOVA)) but not DCs. Data shown are representative of (a) six and (b) two experiments performed. (c) Splenocytes from WT mice (n=4) were stimulated with H. pylori SS1 for 24 h, with or without PAR1-activating peptide and recombinant IL-12 (rIL-12) at 5 or 10 μg ml−1. The inhibitory effects of PAR1 activation on H. pylori–induced secretion of interferon-γ (IFNγ; *ANOVA) was reversed by addition of rIL-12 (#P<0.008 cf H. pylori+PAR1 peptide; ANOVA). In parallel cultures within the same experiment, depletion of macrophages significantly reduced secretion of H. pylori–induced IFNγ (^ANOVA). Box-plots present the median (horizontal bar), interquartile range (box), and 10th and 90th percentiles (bars).
PAR1 suppression of IRF5 expression
Bacterial-induced secretion of IL-12 and IL-23 by macrophages is promoted by the transcription factor interferon regulatory factor 5 (IRF5).22 We thus hypothesized that PAR1 may regulate H. pylori–induced gastritis via IRF5. Analysis of complete gastric tissues revealed no significant difference in IRF5 mRNA levels between uninfected wild-type and PAR1−/− mice. When infected with H. pylori, however, PAR1−/− gastric tissues expressed significantly elevated IRF5 compared with wild-type mice (Figure 8a). A direct comparison of immune cells revealed that PAR1−/− macrophages (Figure 8b) and splenocytes (Figure 8c) have elevated IRF5 even when unstimulated, demonstrating a fundamental role for PAR1 in IRF5 regulation. When wild-type splenocytes were stimulated with H. pylori in vitro, IRF5 expression was upregulated, but importantly, upregulation of IRF5 was completely blocked by the addition of a PAR1-activating but not control peptide (Figure 8d) proving that H. pylori induces IRF5 upregulation in immune cells via a process that is inhibited by PAR1 activation.

Protease-activated receptor 1 (PAR1) regulates interferon regulatory factor 5 (IRF5) expression in both the infected gastric mucosa and immune cells. IRF5 mRNA in PAR1−/− and wild-type (WT) mice and cells was quantified by quantitative PCR (qPCR) in (a) the stomachs of uninfected and Helicobacter pylori–infected mice (n=8), (b) unstimulated peritoneal macrophages (n=4), and (c) unstimulated splenocytes (n=7). Data presented are from one of two similar experiments. PAR1-deficient cells contained significantly increased IRF5 levels, even without stimulation (analysis of variance (ANOVA)). (d) Stimulation of splenocytes from WT mice (n=4) with live H. pylori induced a significant increase in IRF5 (*ANOVA cf unstimulated controls). The addition of PAR1-activating peptide, but not a control peptide, specifically suppressed H. pylori induction of IRF5 to background levels. (e) Gastric macrophages were isolated from the stomach of uninfected (n=3) and 1-week-infected (n=5) WT and PAR1−/− mice by flow cytometry using the parameters described in Figure 3 and IRF5 mRNA levels assessed by qPCR. PAR1−/− gastric macrophages had significantly higher levels of IRF5 (ANOVA). Not significantly different (n.s.).
In order to relate these in vitro observations back to the gastric mucosa, IRF5 levels were quantified in macrophages isolated from the stomachs of uninfected and infected mice. IRF5 levels were higher in PAR1−/− gastric macrophages from both uninfected and infected mice; because of small group sizes, this only reached significance when the groups were pooled (Figure 8).
Finally, we confirmed that regulation of IRF5 mRNA levels in immune cells translated to modified expression of IRF5 protein. Western blot analysis showed that PAR1−/− splenocytes and macrophages both express increased levels of IRF5 protein compared with wild-type cells (Figure 9).

Protease-activated receptor 1 (PAR1) suppresses interferon regulatory factor 5 (IRF5) protein expression in immune cells. Basal IRF5 protein levels relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were assessed by western blotting in (a) splenocytes and (b) peritoneal macrophages. Densitometry values are shown (individual mice and median). PAR1−/− cells express significantly higher levels of IRF5 protein than wild-type (WT) cells (analysis of variance (ANOVA)).
DISCUSSION
Host factors that regulate the severity of H. pylori–induced gastritis likely play a critical role in determining which individuals succumb to associated disease, including gastric adenocarcinoma. In this study we have demonstrated that one such host factor, PAR1, suppresses both Th1- and Th17-immune responses in the gastric mucosa of H. pylori–infected mice via a process involving bone marrow–derived cells. The latter observation was surprising as we had previously shown that PAR1 inhibits the production of the inflammatory cytokine MIP-2 by primary gastric epithelial cells.17 These findings indicate that although PAR1 plays a role in regulating epithelial cytokine production in vitro, during gastric H. pylori infection, it is the PAR1-expressing immune cells that have the dominant effect. In support of this, in vitro analyses of immune cells revealed that PAR1 suppresses Th1 and Th17 responses by inhibiting the secretion of both IL-12 (a key Th1-promoting cytokine) and IL-23 (a key Th17-promoting cytokine) by macrophages.
These data, combined with our previous demonstration of a greatly exacerbated atrophic gastritis in PAR1−/− but not wild-type mice,17 clearly indicate that in the case of H. pylori infection, PAR1 acts to suppress inflammation. However, this is not always the role of PAR1. Although the literature consistently demonstrates an important role for PAR1 in the regulation of inflammation, the nature of that role varies. For example, PAR1 is anti-inflammatory in oxazolone-induced colitis23 and H. pylori infection,17 but proinflammatory in viral (influenza) infection of mice.24 These findings suggest a highly complex role for PAR1 in regulating inflammation and, in particular, indicate that PAR1 can have either pro- or anti-inflammatory activity, depending on the nature of the stimulus and/or the immune cells involved. The latter point is supported by observations that PAR1 is proinflammatory for DCs in a mouse model of endotoxic shock,25 whereas our data presented here clearly show anti-inflammatory activity for PAR1 in macrophages.
Related to this is the effect of PAR1 on vaccination. The mechanism by which vaccines reduce H. pylori colonization in mice is unknown, but is believed to be antibody independent,26, 27 is T-cell mediated, and has been linked to Th17 cells and IL-17,20, 21, 26 although not all evidence supports this.28 Hence, although PAR1 protects against the pathological consequences of H. pylori infection by suppressing the production of proinflammatory cytokines, the same property appears to also reduce the efficacy of vaccine-mediated protection. The increased vaccine efficacy seen in PAR1−/− mice, which when infected have elevated levels of gastric IL-17, is consistent with protection being mediated via this cytokine. However, the effects of PAR1 suppression of other cytokines on vaccine-mediated protection cannot be ruled out.
In support of this possibility, although PAR1 modifies vaccine-mediated protection, there is no evidence that it influences colonization during natural infection in the absence of severe gastritis. We have shown that wild-type and PAR1−/− mice exhibit no difference in H. pylori colonization levels from 1 day to 1 month after infection,17 and this occurs despite elevated IL-17 levels in the gastric mucosa of infected mice (Figure 2). Hence, increased gastric IL-17 levels alone were not sufficient to reduce H. pylori colonization, and vaccination must therefore induce an additional, currently unidentified, response. Colonization does drop in PAR1−/− mice relative to wild-type controls, but only after 2 months of infection and coincident with development of severe gastritis17 that is well known to affect H. pylori numbers in the gastric mucosa.5 Interestingly, although macrophage PAR1 appears to inhibit H. pylori vaccine efficacy, DC PAR2 appears to promote vaccine-mediated protection,29, 30 indicating a complex interaction of these two PAR family members exerting their effects via different immune cells.
The most important discovery relating to the precise mechanism by which PAR1 regulates H. pylori pathogenesis is the finding that this receptor not only suppresses expression of the important transcription factor IRF5 in the infected gastric mucosa, but that it also regulates IRF5 expression by immune cells. It is notable that PAR1−/− macrophages express extremely high levels of IRF5 compared with other cells (Figure 8b,c), and that PAR1−/− macrophages isolated from the gastric mucosa express more IRF5 than wild-type macrophages (Figure 8e). This observation, combined with our demonstration of significantly higher macrophage numbers in the gastric mucosa of H. pylori–infected PAR1−/− compared with wild-type mice (Figure 3), suggests that the elevated levels of IRF5 observed in the infected PAR1−/− stomach (Figure 8a) are the result of gastric macrophages.
As IRF5 is a key promoter of IL-12 and IL-23 secretion and thus Th1 and Th17 immunity,22 our finding that PAR1 is an important regulator of IRF5 provides a mechanism for the suppression of inflammation by this receptor. Based on our data, we propose a model by which H. pylori infection activates IRF5 (as shown in Figure 8d) that then drive macrophages to secrete IL-12 and IL-23 (Figure 7b and c); these cytokines then promote the development of a mixed Th1 and Th17 immune response (Figures 2 and 6). H. pylori infection has been shown to increase thrombin levels in the gastric mucosa.31 Thrombin (considered the most important PAR1 activating serine protease)14 would then activate PAR1 on macrophages, causing reduced IRF5 expression, and Th1 and Th17 responses, resulting in a suppressed gastritis.
In conclusion, PAR1 suppression of IRF5 and IL-12/23 secretion by macrophages provides a novel mechanism by which the host can suppress Th1 and Th17 response to H. pylori and thereby protect itself against the pathological consequences of this infection. Dysregulation of this process may be an important factor in the susceptibility of some individuals to H. pylori–associated disease, and may explain the association of a PAR1 polymorphism (PAR1 IVSn −14 A/T; known to affect PAR1 activity) with susceptibility to H. pylori–associated gastric cancer.19
METHODS
Bacterial culture. For infection of mice and cell stimulation assays, SS1, H. pylori strain SS1,32 was cultivated in brain heart infusion broth (BHI; Oxoid, Basingstoke, UK) containing 5% horse serum (JRH Biosciences, Lenexa, KS), 0.02% amphotericin B, and Skirrow’s Selective Supplements under microaerophilic conditions for 24 h at 37 °C. For formalin fixation for vaccination,33 bacteria were suspended in 0.01 M formaldehyde in phosphate-buffered saline (PBS) and adjusted to an OD600 of 1.5. After 2 h of gentle shaking at 37 °C, the bacteria were shaken overnight at room temperature, washed 3 times in PBS, and resuspended at 108 bacteria per ml.
Infection of mice. PAR1−/−(see ref. 34) and wild-type specific-pathogen free C57BL/6 mice were bred within the Veterinary Science animal house, University of Melbourne (Parkville, Melbourne, Australia). All experiments involved age-matched female mice and were performed under University of Melbourne Animal Ethics Committee approval. Mice were infected intragastrically once with 107 H. pylori suspended in 0.1 ml BHI.
Generation of radiation chimeras. Recipient mice were irradiated with 2 doses of 550 rads, 3 h apart, using a Co60 source (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia). The next day, bone marrow cells were harvested from the femurs and tibias of donor mice in PBS with 1% fetal calf serum (FCS; HyClone, Logan, UT). Cells were washed once, and then 2 × 106 cells were injected into recipients intravenously in PBS. Mice were maintained on drinking water containing 2 mg ml−1 neomycin (Gibco, Basel, Switzerland) for 2 weeks. Mice were left for 2 months before infection to allow reconstitution.
Quantification of H. pylori colonization by colony-forming assay. H. pylori infection levels within mouse gastric tissues were quantified by colony-forming assay. Briefly, stomachs were opened along the inner curvature and divided into two halves. One half was placed in BHI and homogenized (GmbH Polytron homogenizer, Kinematica, Lucern, Switzerland). Tenfold serial dilutions were prepared in BHI broth and aliquots spread over GSSA selective agar plates (Blood Agar Base No. 2 with 5% horse blood, vancomycin hydrochloride (12 μg ml−1), polymyxin B (0.4 μg ml−1), bacitracin (24 μg ml−1), nalidixic acid (1.3 μg ml−1), and amphotericin B (5 mg ml−1)). After 5 days of culture as above, colonies were counted and the number of colony-forming units calculated per stomach.35
Histological assessment of gastritis. Gastritis was assessed histologically as described previously.36 Briefly, stomach halves were fixed in 10% neutral buffered formalin, embedded in paraffin, and 4 μm thick sections stained with hematoxylin and eosin and scored blinded under light microscopy. Inflammation was assessed as: (i) cellular infiltration (lymphocytes and neutrophils) into the lamina propria, graded from 0 to 6 (0=none; 1=mild multifocal; 2=mild widespread or moderate multifocal; 3=mild widespread and moderate multifocal, or severe multifocal; 4=moderate widespread; 5=moderate widespread and severe multifocal; 6=severe widespread); and (ii) atrophy (loss of parietal cells), graded from 0 to 3 (absent, mild, moderate, severe).
Flow cytometry. Gastric cellular infiltrate to be analyzed by flow cytometry were isolated by perfusion using a technique modified from Alderuccio et al.37 Stomachs were collected into Hanks’ balanced salt solution (HBSS; Gibco), perfused with HBSS containing 5% FCS (Gibco), 5 mM EDTA, and 1 mM dithiothreitol (perfusion solution), and incubated at 37 °C for 15 min and then cut into small pieces and poured through a 70 μm cell strainer. After a further 15 min in 10 ml of perfusion solution, pieces were vortexed and again poured through a 70 μm cell strainer. Cells in both filtrates were pooled and collected by centrifugation at 600 g for 5 min. The cell pellet was resuspended in 4 ml of 20% Percoll (Sigma, St Louis, MO), and then overlaid on a 70%/40% Percoll discontinuous gradient and centrifuged at 600 g for 10 min. The interphase was collected, washed with 2% FCS in PBS, and blocked in 100 μl of 20% mouse serum (collected in-house) and 1 μg/ml anti-FcγII/FcγIII (2.4G2, Walter and Eliza Hall Institute) on ice for 20 min. Cells were stained with anti-MHCII-FITC (M5/114.15.2; Caltag Laboratories, Burlingame, CA), anti-CD45-PE.Cy5.5 (30-F11; BioLegend, San Diego, CA), anti-CD11b-Brilliant Violet 421 (M1/70; BioLegend), anti-CD19-Brilliant Violet 510 (6D5; BioLegend), anti-CD4-Brilliant Violet 650 (RM4-5; BioLegend), and anti-Ly6G-APC.Cy7 (1A8; BioLegend). Cells were washed twice, resuspended in 0.25 mg/ml propidium iodide (ImmunoChemistry Technologies, Bloomington, MN) to exclude dead cells, and acquired on a BD Influx cell sorter (BD Biosciences, San Jose, CA). Cell counts were normalized using AccuCount Fluorescent Particles (Spherotech, Lake Forest, IL). Data were analyzed using FCS Express (De Novo Software, Los Angeles, CA).
Cell stimulation assays. Peritoneal macrophages and bone marrow–derived DCs were prepared as described previously.17 Briefly, noninduced peritoneal cells obtained by sterile lavage with ice-cold PBS supplemented with 1% (v/v) FCS were washed, and resuspended in RPMI-1640 with 10% FCS (Gibco, complete RPMI). Peritoneal macrophages were enriched to >95% purity by adherence to tissue culture plastic for 1 h, with nonadherent cells removed by washing. Red cell–depleted splenocytes, macrophages, and bone marrow–derived DCs were cultured at 106 cells per ml in complete RPMI with 100 μM TFLLR PAR1 agonist peptide or FTLLR control peptide (Auspep, Tullamarine, Victoria, Australia) for 10 min before addition of H. pylori (multiplicity of infection of 10). Macrophages were depleted from splenocytes as described above for peritoneal macrophage enrichment. Recombinant IL-12 was purchased from eBioscience (San Diego, CA).
Quantification of cytokine levels by ELISA. Half stomachs and whole mesenteric lymph nodes were homogenized in PBS. Culture supernatants and homogenates were centrifuged to remove debris before enzyme-linked immunosorbent assay (ELISA). The 96-well Maxisorp plates (Nunc, Roskilde, Denmark) were coated overnight with anti-mouse IFNγ (0.1 μg per well) or IL-10 (0.05 μg per well; BD Biosciences); IL-17A (0.5 μg per well), IL-23 (0.1 μg per well), transforming growth factor-β (0.1 μg per well) or IL-13 (0.5 μg per well; eBioscience) or MIP-2 (0.1 μg per well; R&D Systems, Minneapolis, MN) in bicarbonate coating buffer, pH 9.6. Plates were blocked with 1% BSA (Sigma) in PBS (blocker) for 1 h before addition of samples in duplicate for 3 h at room temperature or 4 °C overnight. Captured cytokines were then labeled with biotinylated anti-mouse IFNγ (0.05 μg per well) or IL-10 (0.05 μg per well; BD Biosciences); IL-17A (0.025 μg per well), IL-23 (0.05 μg per well), transforming growth factor-β (0.05 μg per well), or IL-13 (0.5 μg per well; eBioscience); MIP-2 (0.1 μg per well; R&D Systems) in blocker for 1 h before the addition of 50 μl horseradish peroxidase–conjugated streptavidin (Pierce, Rockford, IL) 1:5,000 in blocker for 30 min. Color was developed with 100 μl of TMB solution prepared as 0.1% of 10 mg ml−1 TMB (Sigma) in dimethyl sulfoxide and 0.006% hydrogen peroxide in phosphate-citrate buffer, pH 5.0, and the reaction stopped with an equal volume of 2 M sulfuric acid before reading absorbance at 450 nm. Sample concentration was determined against a standard curve of respective recombinant cytokines (same manufacturers as antibodies). IL-12p70 was quantified by ELISA kit (R&D Systems) as per the manufacturer’s instructions.
Quantification of antibody response to infection. Sera were collected by cardiac puncture. Intestinal mucus scrapings were collected from the lower 10 cm of the longitudinally opened small intestine using a scalpel blade, weighed, and then mixed with an equal volume of PBS containing complete mini-EDTA-free proteinase inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Anti-Helicobacter antibody levels were determined by direct ELISA. Maxisorp immunoplates (Nunc) were coated overnight with 50 μl of H. pylori lysate (100 μg ml−1) in bicarbonate buffer, and then blocked as above for 45 min at room temperature. Samples were serially diluted 1:10 in blocker and 50 μl added to duplicate wells before incubation at room temperature for 1 h. After washing, 50 μl of either horseradish peroxidase–conjugated goat-anti-mouse IgG1 (Southern Biotech, Birmingham, AL; diluted 1:6,000), IgG2c (Immunology Consultants Laboratory, Newberg, OR; diluted 1:5,000), or IgA (Southern Biotech; diluted 1:5,000) in blocker were added per well and incubated at room temperature for 45 min. Color was developed by addition of TMB solution, and the reaction stopped by adding sulfuric acid as above. Absorbance was read at 450 nm and end point titers calculated.
Vaccination against H. pylori. Mice (n=8) were dosed orogastrically with 200 μl of either (i) PBS or (ii) 107 formalin-fixed H. pylori SS1 mixed with 20 μg UEA-I.38 Mice received two vaccinations 3 weeks apart. At 4 weeks after the second vaccination, all mice were challenged with a single orogastric dose of 107 H. pylori SS1 in 100 μl of BHI broth. At 4 weeks after challenge, H. pylori infection levels within mouse gastric tissues were quantified by colony-forming assay as above.
Quantification of IRF5. RNA from gastric tissues and cultured cells was extracted using Tri Reagent (Ambion, Austin, TX) according to the manufacturer’s instructions. RNA from FACS-sorted cells was extracted by disrupting in buffer RLT by passing through a 27-G syringe, adding 640 μg of carrier RNA (Qiagen, Venlo, The Netherlands), and then using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Complementary DNA was transcribed using the Quantitect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions.
IRF5 mRNA expression levels in gastric tissues and cells were quantified by PCR. Primers for murine Actb and IRF5 were as previously described.39, 40 The reaction was performed in duplicate in 25 μl containing 2 μl complementary DNA, 0.2 μM primers, and 12.5 μl 2 × SYBR Green PCR Mastermix (Promega, Madison, WI) using an Mx3005P cycler (Stratagene, La Jolla, CA). Cycling conditions were as follows: 1 cycle at 94 °C for 5 min, 40 cycles at 94 °C for 30 s, 45 cycles at 60 °C for 30 s, and 40 cycles at 72 °C for 30 s. Expression was determined by the REST formula,41 relative to β-actin.
For western blot, macrophages were lysed in radioimmunoprecipitation buffer (150 mmol/l NaCl, 1% IGEPAL, 0.5% deoxycholic acid, 0.1% SDS, 50 mmol l−1 Tris, pH 7.5) containing protease inhibitor cocktail (Roche). Protein samples were resolved on a denaturing 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham Biosciences, Amersham, UK). The membrane was blocked with PBS containing 5% skim milk and 0.05% Tween-20 (Sigma) and then probed with anti-IRF5 monoclonal antibody (1:500, Abcam, Cambridge, UK) and anti-GAPDH (1:1,000, Cell Signaling Technology, Danvers, MA). The primary antibodies were detected with horseradish peroxidase–conjugated anti-rabbit antibody (1:1,000, Dako, Glostrup, Denmark). Blots were developed with ECL Prime Detection Reagent (Amersham Biosciences) and visualized using an ImageQuant LAS 4000 (GE Healthcare, Fairfield, CT, USA). Densitometry was analyzed using ImageJ (US National Institutes of Health, Bethesda, MD).
Statistics. Statistical analyses were performed using SPSS software, version 21.0 (IBM, Armonk, NY). Histological grading scores were compared by nonparametric Mann–Whitney analysis. All other data were log-transformed and compared by one-way analysis of variance with Dunnett’s post hoc analysis where required.
References
Uemura, N. et al. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 345, 784–789 (2001).
Yuan, Y., Padol, I.T. & Hunt, R.H. Peptic ulcer disease today. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 80–89 (2006).
Shiomi, S. et al. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter 13, 518–524 (2008).
Bamford, K.B. et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 114, 482–492 (1998).
Eaton, K.A., Mefford, M. & Thevenot, T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J. Immunol. 166, 7456–7461 (2001).
Kimang'a, A. et al. IL-17A and IL-17F gene expression is strongly induced in the mucosa of H. pylori-infected subjects from Kenya and Germany. Scand. J. Immunol. 72, 522–528 (2010).
Kaparakis, M. et al. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect. Immun. 76, 2235–2239 (2008).
Tegtmeyer, N., Wessler, S. & Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 278, 1190–1202 (2011).
Vinall, L.E. et al. Altered expression and allelic association of the hypervariable membrane mucin MUC1 in Helicobacter pylori gastritis. Gastroenterology 123, 41–49 (2002).
McGuckin, M.A. et al. Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis. Gastroenterology 133, 1210–1218 (2007).
El-Omar, E.M. et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404, 398–402 (2000).
Sugimoto, M. et al. Different effects of polymorphisms of tumor necrosis factor-alpha and interleukin-1 beta on development of peptic ulcer and gastric cancer. J. Gastroenterol. Hepatol. 22, 51–59 (2007).
Dery, O., Corvera, C.U., Steinhoff, M. & Bunnett, N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 274 (6 Pt 1), C1429–C1452 (1998).
Soh, U.J., Dores, M.R., Chen, B. & Trejo, J. Signal transduction by protease-activated receptors. Br. J. Pharmacol. 160, 191–203 (2010).
Sekiguchi, F., Takaoka, K. & Kawabata, A. Proteinase-activated receptors in the gastrointestinal system: a functional linkage to prostanoids. Inflammopharmacology 15, 246–251 (2007).
Shpacovitch, V., Feld, M., Bunnett, N.W. & Steinhoff, M. Protease-activated receptors: novel PARtners in innate immunity. Trends Immunol. 28, 541–550 (2007).
Wee, J.L. et al. Protease-activated receptor-1 down-regulates the murine inflammatory and humoral response to Helicobacter pylori. Gastroenterology 138, 573–582 (2010).
Lurje, G. et al. Genetic variations in angiogenesis pathway genes associated with clinical outcome in localized gastric adenocarcinoma. Ann. Oncol. 21, 78–86 (2010).
Castano-Rodriguez, N. et al. PAR-1 polymorphisms and risk of Helicobacter pylori-related gastric cancer in a Chinese population. Anticancer Res. 32, 3715–3721 (2012).
DeLyria, E.S., Redline, R.W. & Blanchard, T.G. Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136, 247–256 (2009).
Velin, D. et al. Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136, 2237–2246 e2231 (2009).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 (2011).
Cenac, N. et al. Proteinase-activated receptor-1 is an anti-inflammatory signal for colitis mediated by a type 2 immune response. Inflamm. Bowel Dis. 11, 792–798 (2005).
Khoufache, K. et al. PAR1 contributes to influenza A virus pathogenicity in mice. J. Clin. Invest. 123, 206–214 (2013).
Niessen, F. et al. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452, 654–658 (2008).
Ermak, T.H. et al. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J. Exp. Med. 188, 2277–2288 (1998).
Sutton, P., Wilson, J., Kosaka, T., Wolowczuk, I. & Lee, A. Therapeutic immunization against Helicobacter pylori infection in the absence of antibodies. Immunol. Cell Biol. 78, 28–30 (2000).
Delyria, E.S. et al. Vaccine-induced immunity against Helicobacter pylori in the absence of IL-17A. Helicobacter 16, 169–178 (2011).
Velin, D. et al. PAR2 promotes vaccine-induced protection against Helicobacter infection in mice. Gastroenterology 141, 1273–1282 1282 e1271 (2011).
Sutton, P. Vaccinating against Helicobacter pylori: dissecting the mechanism. Gastroenterology 141, 1149–1151 (2011).
Suzuki, Y. et al. Vacuolating cytotoxin A is associated with increased thrombin generation in gastric mucosa. Helicobacter 10, 323–331 (2005).
Lee, A., O'Rourke, J., De Ungria, M.C., Robertson, B., Daskalopoulos, G. & Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112, 1386–1397 (1997).
Raghavan, S., Hjulstrom, M., Holmgren, J. & Svennerholm, A.M. Protection against experimental Helicobacter pylori infection after immunization with inactivated H. pylori whole-cell vaccines. Infect. Immun. 70, 6383–6388 (2002).
Connolly, A.J., Ishihara, H., Kahn, M.L., Farese, R.V. Jr. & Coughlin, S.R. Role of the thrombin receptor in development and evidence for a second receptor. Nature 381, 516–519 (1996).
Sutton, P. Considering increased mouse stomach mass, when calculating prophylactic vaccine efficacy against Helicobacter pylori. Helicobacter 12, 210–212 (2007).
Sutton, P. et al. Post-immunisation gastritis and Helicobacter infection in the mouse: a long term study. Gut 49, 467–473 (2001).
Alderuccio, F., Toh, B.H., Gleeson, P.A. & van Driel, I.R. A novel method for isolating mononuclear cells from the stomachs of mice with experimental autoimmune gastritis. Autoimmunity 21, 215–221 (1995).
Chionh, Y.T., Wee, J.L., Every, A.L., Ng, G.Z. & Sutton, P. M-cell targeting of whole killed bacteria induces protective immunity against gastrointestinal pathogens. Infect. Immun. 77, 2962–2970 (2009).
Rad, R. et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131, 525–537 (2006).
Yanai, H. et al. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. USA 104, 3402–3407 (2007).
Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
Acknowledgements
This work was supported by the Victorian Government’s Operational Infrastructure Support Program and by Project grant 508963 from the National Health and Medical Research Council of Australia. P.S. is supported by a Senior Research Fellowship from the NHMRC. We gratefully acknowledge the assistance of Matthew Burton with flow cytometric analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Chionh, YT., Ng, G., Ong, L. et al. Protease-activated receptor 1 suppresses Helicobacter pylori gastritis via the inhibition of macrophage cytokine secretion and interferon regulatory factor 5. Mucosal Immunol 8, 68–79 (2015). https://doi.org/10.1038/mi.2014.43
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.43
This article is cited by
-
The TLR/MyD88 signalling cascade in inflammation and gastric cancer: the immune regulatory network of Helicobacter pylori
Journal of Molecular Medicine (2023)
-
Identification of new regulatory genes through expression pattern analysis of a global RNA-seq dataset from a Helicobacter pylori co-culture system
Scientific Reports (2020)
-
The MUC1 mucin specifically inhibits activation of the NLRP3 inflammasome
Genes & Immunity (2016)
