
Abstract
Entamoeba histolytica (Eh) is an extracellular protozoan parasite of the human colon, which occasionally breaches the intestinal barrier. Eradicating ameba that invades is essential for host survival. A defining but uncharacterized feature of amebic invasion is direct contact between ameba and host cells. This event corresponds with a massive pro-inflammatory response. To date, pathogen recognition receptors (PRRs) that are activated by contact with viable Eh are unknown. Here we show that the innate immune system responds in a qualitatively different way to contact with viable Eh vs. soluble ligands produced by viable or dead ameba. This unique Eh Gal-lectin contact-dependent response in macrophages was mediated by activation of the inflammasome. Soluble native Gal-lectin did not induce inflammasome activation, but was sufficient for transcriptional priming of the inflammasome and non-inflammasome-dependent pro-inflammatory cytokine release. We conclude the inflammasome is a pathogenicity sensor for invasive Eh and identify for the first time a PRR that specifically responds to contact with intact parasites in a manner that accords with scale immune response to parasite invasion.
Similar content being viewed by others


Introduction
Entamoeba histolytica (Eh) is an extracellular protozoan parasite of humans that either harmlessly colonizes the mucus layer of the colon or triggers disease when it moves from the intestinal lumen into sterile tissues and elicits an aggressive inflammatory response. Invasive infections may lead to acute amebic dysentery, chronic amebic colitis, or disseminated extraintestinal infections that produce amebic abscesses, which annually cause 100 million cases of amebiasis leading to 100,000 deaths.1
The pathogenesis of amebiasis involves destruction of the intestinal mucus layer, followed by direct contact and adherence of ameba to host cells.2 It has been a longstanding observation that the quality and magnitude of the host response is different during contact compared with when ameba are sensed offshore.3, 4 Intuitively this makes sense; upon sensing microbes innate immune cells are faced with the task of determining the level of danger to the host and initiating appropriate antimicrobial responses. Ameba that are directly in contact with host tissues present a more imminent danger than ameba located further away, necessitating a rapid and vigorous response precisely at the site of infection. Furthermore, some host defenses are beneficial only during microbial invasion and are tightly regulated to prevent unnecessary injury to the host. The pathogen recognition receptors (PRRs) and cellular responses selectively activated as a result of direct contact with Eh are unknown.
Previous studies have reported that interleukin (IL)-1β is released during invasion, which involves sequential disruption of the colonic epithelial barrier leading to direct contact of ameba with the underlying lamina propria, where resident and recruited myeloid cells are present that are the major producers of IL-1β.5, 6, 7 As inflammasomes are central regulators of IL-1β processing and secretion, this suggests that direct sensing of Eh might induce inflammasome activation leading to the release of IL-1β and a severe inflammatory response aimed at removing ameba from the tissue. Inflammasomes are cytosolic sensors of pathogens and various cellular stresses that upon activation form high molecular weight complexes that mediate recruitment and activation of caspase-1.8 Active caspase-1 in turn cleaves the precursors of IL-1β and IL-18 into bioactive fragments and mediates their release along with several other leaderless proteins via an undefined casaspe-1-dependent secretion event.8, 9 In this study, we sought to determine whether Eh provokes inflammasome activation via an adherence-dependent mechanism.
Adherence of Eh during both colonization and invasion is directly mediated by a 170-kDa surface Gal/GalNAc lectin (Gal-lectin), which binds galactose and N-acetylgalactosamine residues on mucins comprising the colonic mucus layer and unknown host cell receptors, respectively.10, 11 Studies in humans and mice have demonstrated Gal-lectin has a critical role activating anti-Eh defenses and is a major target of adaptive immunity.12, 13, 14, 15, 16 PRRs activated by the Gal-lectin are, however, unknown. Our studies reveal that Eh induces inflammasome activation through a contact-dependent event involving binding of surface-associated Gal-lectin to the macrophage membrane, leading to release of IL-1β, IL-18, and several additional cytokines that require active caspase-1 for secretion. In contrast to surface-associated Gal-lectin, soluble Gal-lectin, which is continually shed from ameba surfaces, induced expression of pro-IL-1β and the inflammasome machinery in naive macrophages, and provoked release of inflammasome-independent pro-inflammatory cytokines. However, soluble Gal-lectin did not induce inflammasome activation. This reveals that attachment of Gal-lectin to host cells has an important role in conveying critical information about the nature of the microbial encounter. In the context of an intact ameba, surface-bound Gal-lectin functions in activating a strong pro-inflammatory response via a contact-dependent event that triggers inflammasome activation and IL-1β release. Alternatively, when ameba are sensed at a distance, soluble Gal-lectin shed from the ameba surface, elicits a weaker inflammatory cytokine response. Thus, during contact the inflammasome is a sensor for detecting direct interactions with microbial surfaces, and therefore selectively senses invasive microbes leading to augmentation of certain host defenses at sites of invasive infection.
Results
E. histolytica induces inflammasome activation via direct contact
To understand how host cells respond differently to ameba in contact and non-contact modes, we first sought to define cytokine signatures of innate immune cells in response to direct contact with ameba and soluble ameba ligands that are detected when ameba are sensed from a distance. For these studies, we used macrophages because they are critical during innate responses to Eh, are primary sources of IL-1β, and other pro-inflammatory mediators, and highly express inflammasomes. Multiplex cytokine array was used to compare cytokine profiles in supernatants of differentiated human THP-1 macrophages stimulated directly with ameba or concentrated secreted components from viable ameba. Direct contact distinctly induced IL-1β, IL-1α, fibroblast growth factor (FGF)-2, macrophage-derived chemokine (MDC), and interferon-γ-induced protein (IP)-10 (Figures 1a–c, Supplementary Figures S1a and b online). In addition to activating IL-1β and IL-18, active caspase-1 is known to regulate IL-1α and FGF-2 secretion. This suggested that direct contact with macrophages induced inflammasome activation. In vivo we also observe high levels of serum IL-1β and IL-18 in gerbils with amebic liver abscess (Supplementary Figures S2a and b). At this stage of infection macrophages, which line the margin of the inner granuloma wall, are the chief cells directly in contact with ameba.17

Entamoeba histolytica (Eh) in direct contact with macrophages induces the secretion of caspase-1-regulated cytokines. (a-c) PMA-differentiated THP-1 macrophages were stimulated with the indicated number of live ameba (Eh) or secreted components from live ameba for 3 h and cytokine secretion was measured by multiplex cytokine array. Live ameba selectively induced secretion of interleukin (IL)-1β (a), IL-1α (b), and fibroblast growth factor (FGF)-2 (c). (d) PMA-differentiated THP-1 macrophages were stimulated with increasing numbers of live ameba for 30 min. Secretion of processed caspase-1 (p35 subunit, p20/p22 subunit and N-terminal CARD domain) and active IL-1β (p17) were detected in cell supernatants (SN) by immunoblot. Total IL-1β (e) and IL-18 (f) secretion were measured by enzyme-linked immunosorbent assay (ELISA). (g) Lipopolysaccharide (LPS)-primed mouse bone marrow-derived macrophages (BDMs) were stimulated with increasing numbers of live ameba for 30 min. Secretion of processed caspase-1 (p10 subunit) and active IL-1β (p17) were detected in cell SNs by immunoblot. ***P<0.005 vs. negative control. n=4.
To determine whether Eh activates an inflammasome, we assessed processing and secretion of caspase-1, and its substrates IL-1β and IL-18 in macrophages stimulated with ameba in contact and non-contact modes. In unstimulated macrophages, caspase-1 exists as an inactive pro-enzyme, and after recruitment to an active inflammasome platform undergoes sequential autoproteolysis that removes the N-terminal CARD and forms an active heterodimer of p10 and p20/22 subunits. Fragments of processed caspase-1 can be detected in cell supernatants upon inflammasome activation, along with active forms of IL-1β and IL-18. Viable ameba in direct contact with differentiated THP-1 macrophages induced rapid dose-dependent processing and secretion of the partially processed p35 form, the active p20/22 subunit, and N-terminal CARD of caspase-1 (Figure 1d). The 26-kDa band on the caspase-1 p20/22 immunoblot is active caspase-4, which is also induced by Eh (unpublished observations). Upon Eh contact, pro-IL-1β underwent cleavage into its active 17-kDa form and was secreted along with IL-18 (Figures 1d–f). Similarly, primed bone marrow-derived macrophages (BDMs) challenged with viable ameba processed and secreted caspase-1 (the mouse anti-caspase-1 antibody used detects the active p10 subunit) and released active IL-1β (Figure 1g). Cell death did not occur, as measured by Trypan blue exclusion assay and lactate dehydrogenase release, ruling out direct cytolytic activity by Eh as a source of cytokine and caspase-1 release in the cell supernatants (data not shown).
To assess whether IL-1β and IL-18 release were caspase-1-dependent, differentiated THP-1 macrophages were pre-treated with the pan-caspase inhibitor zVAD-fmk and the caspase-1-specific inhibitor zYVAD-fmk. Caspase-1 inhibition prevented release of active 17 kDa IL-1β and IL-18 (Figures 2a–c). Secretion of IL-1α and FGF-2 was also sensitive to caspase-1 inhibition (Supplementary Figures S3a and b). These results were confirmed in caspase-1−/− BDMs, which did not process and release IL-1β in response to Eh (Figure 2d). Thus, secretion of IL-1β, IL-18, and the other contact-dependent cytokines was an inflammasome-regulated event. In contrast to direct contact, we did not observe inflammasome activation by any non-contact modes, which included lysed ameba, ameba membrane components, ameba cytosolic components, viable ameba separated on transwells, and concentrated secreted components from viable ameba (Figures 3a–d). These data were consistent with the cytokine array that revealed contact specifically induced cytokines that are regulated by caspase-1 (Figures 1a–c).

Contact-dependent cytokine secretion induced by Entamoeba histolytica (Eh) is regulated by the inflammasome. PMA-differentiated THP-1 macrophages were pre-treated for 60 min with zVAD-fmk (100 μM) or YVAD-fMK (100 μM) and stimulated with 1.5 × 103 live ameba for 30 min. (a) Secretion of processed caspase-1 (p35 subunit, p20/p22 subunit and N-terminal CARD domain) and active interleukin (IL)-1β (p17) were detected in cell supernatants (SN) by immunoblot. Total IL-1β (b) and IL-18 (c) secretion were measured by ELISA. (d) Lipopolysaccharide (LPS)-primed Wt and caspase-1−/− bone marrow-derived macrophages (BDMs) were stimulated with 1.5 × 103 live ameba or nigericin (10 μM) for 30 min and secretion of processed caspase-1 (p10 subunit) and active IL-1β (p17) were detected in cell SNs by immunoblot. ***P<0.005 vs. negative control. n=4.

Inflammasome activation by Entamoeba histolytica (Eh) requires intact ameba and direct contact. (a, b) PMA-differentiated THP-1 macrophages were exposed to 1.5 × 103 live ameba or equivalent amounts of concentrated secretory components, whole ameba lysate, ameba membrane fraction, or ameba cytosolic fraction for 30 min. (c, d) PMA-differentiated THP-1 macrophages were exposed to 1.5 × 103 live ameba for 30 min or the indicated number of live ameba placed on the top chamber of transwells for 2 h. (a, c) Secretion of processed caspase-1 (p35 subunit, p20/p22 subunit and N-terminal CARD domain) was detected in cell supernatants (SN) by immunoblot. (b, d) Total interleukin (IL)-1β secretion was measured by enzyme-linked immunosorbent assay (ELISA). ***P<0.005 vs. negative control. n=4.
To further confirm this observation, we used a caspase-1-specific fluorescence-labeled inhibitor of caspases, the YVAD-FLICA, to visualize localization of active capase-1 in contact and non-contact modes with ameba (Figures 4a and b). Ameba that directly contacted macrophages induced intense staining of the YVAD-FLICA throughout the cytoplasm, which was similar to induction with a rapid inflammasome stimulus, lipopolysaccharide (LPS) and nigericin (Figure 4a). The intensity of active caspase-1 though, was much greater in the cytoplasm after ameba challenge compared with LPS and nigericin. This could be due to rapid loss of membrane integrity that occurs with LPS nigericin resulting in less caspase-1 retention in the cytoplasm. Consistent with this, we observed that macrophages contacted by ameba retained membrane integrity and did not undergo caspase-1-dependent cell death, even after several hours. It has been recently reported18 that distinct caspase-1-containing complexes perform cell death and cytokine processing, and may explain why Eh induces rapid cytokine release without causing cell death. During the initial interaction with ameba, intense caspase-1 activation occurred around the plasma membrane that formed the contact site with ameba, suggesting the inflammasome was triggered by a direct interaction between the Eh and macrophage membrane (Figure 4a bottom panel macrophage located at the top of image). The YVAD-FLICA did not label macrophages that were untouched by ameba, confirming on a single-cell basis that inflammasome activation by Eh is contact dependent. Additionally, the YVAD-FLICA also stained ameba that triggered inflammasome activation (Figure 4a). The YVAD-FLICA, however, did not label ameba cultured without macrophages or ameba that are inhibited from activating the inflammasome. The staining may be from the uptake of active caspase-1 from culture supernatants or leakage of active caspase-1 from phagosomes containing inflammasome-activated macrophages. Alternately, Eh may have an enzyme that is activated in response to contact with macrophages and binds the YVAD sequence. Interestingly, while investigating active caspase-1 with the YVAD-FLICA, we observed ameba ingesting intact macrophages resulting in numerous giant amebas packed with macrophages, which were evident by the colocalization of 4,6-diamidino-2-phenylindole (does not label ameba DNA) and intense YVAD-FLICA foci inside the ameba (Figures 4a and b). During Eh invasion, we suspect that ingestion of intact macrophages could be a strategy by the parasite to dampen the intensity of the inflammatory response, as it would certainly limit the duration of inflammasome activation.

Direct Entamoeba histolytica (Eh) contact recruits active caspase-1 to the cell surface. (a, b) Confocal images of PMA-differentiated THP-1 macrophages showing caspase-1 activation after 30 min of exposure to live Eh or nigericin (10 μM) in which active caspase-1 were stained with an YVAD-florescent-labeled inhibitor of caspase (FLICA). Mammalian nuclei were labeled with 4,6-diamidino-2-phenylindole (DAPI; ameba nuclei are not stained by DAPI). Arrowhead points to contact between the macrophage and ameba surface. Note that active caspase-1 accumulates around the contact surface. Arrows point to macrophages that have been phagoyctosed by an ameba. (b) An ameba that ingested several macrophages. Scale bar=20 μm, Mφ, macrophage.
Surface-bound Gal-lectin is required for inflammasome activation
As contact was clearly necessary for Eh to activate the inflammasome, this indicated adherence to the macrophage plasma membrane may be required. We12, 19 have previously shown that human immune sera following invasive amebiasis contains potent adherence-inhibitory antibodies that prevent attachment of ameba to cells and intestinal mucin the critical receptor for Eh to establish colonization of the colon. In our assay, human immune sera dose-dependently inhibited processing and secretion of caspase-1 and IL-1β in response to ameba (Figures 5a–c). No inhibition occurred in the presence of normal human sera, indicating that amebic adherence via a surface-bound molecule-mediated inflammasome activation (Figures 5a–c). Similar results were obtained for IL-18, except that normal human sera augmented IL-18 release in response to ameba. Importantly though, this was significantly inhibited by immune human sera (Figure 5d). In a similar manner, IL-1α, FGF-2, and MDC secretion was also inhibited by human immune serum (Supplementary Figures S4a–c).

Serum of patients with invasive amebiasis inhibits Entamoeba histolytica (Eh)-induced inflammasome activation. (a-d) Ameba were treated with pooled human immune sera of patients with amebic liver abscess or normal immune serum at 1:100 or the indicated dilution for 10 min at 4 °C before incubation with PMA-differentiated THP-1 macrophages for 30 min. (a) Secretion of processed caspase-1 (p35 subunit, p20/p22 subunit and N-terminal CARD domain) and (b) active interleukin (IL)-1β (p17) were detected in cell supernatants (SN) by immunoblot. Total IL-1β (c) and IL-18 (d) secretion were measured by enzyme-linked immunosorbent assay (ELISA). (e) Human immune serum was used to detect the 170 and 130 kDa forms of Eh Gal/GalNAc lectin (Gal-lectin; 1 μg per lane) or in the total lysates of Eh. ***P<0.005 vs. negative control. n=4.
Following invasive amebiasis, human serum contains high levels of antibodies against the antigenically conserved Eh Gal-lectin. Human immune sera recognized the purified 170 kDa Gal-lectin and surface-bound Gal-lectin in the lysates of whole ameba (Figure 5e).20 As Gal-lectin is abundantly expressed on ameba membranes and is a major adherence molecule, we sought to determine if contact-dependent inflammasome activation required Gal-lectin. The binding activity of Gal-lectin is inhibited in the presence of galactose monomers.10, 11 We found that addition of galactose at a concentration that maximally inhibits Gal-lectin binding during exposure of differentiated THP-1 cells to ameba completely abolished caspase-1 and IL-1β processing and secretion (Figures 6a and b). Similarly, secretion of IL-18, IL-1α, FGF-2, and MDC was also abolished (Figure 6c, Supplementary Figure 5a–c). Glucose used an osmotic control, did not alter caspase-1 and IL-1β activation or the secretion of the other inflammasome-regulated cytokines. Furthermore, galactose inhibition achieved the same results in BDMs, suggesting the role of Gal-lectin in activating the inflammasome is conserved (Figure 6d). Glutaraldehyde treatment inactivated Gal-lectin binding, and like galactose inhibition, ameba fixed in glutaraldehyde (1%) did not trigger the inflammasome (Supplementary Figures S5d and e).11, 21 We also tested whether human intestinal mucin, which is the only identified host receptor that binds with high affinity to the Gal-lectin, could competitively inhibit the inflammasome.11 As predicted, ameba pre-treated with purified human colonic mucin dose-dependently inhibited processing and secretion of caspase-1 and IL-1β in differentiated THP-1 macrophages (Figures 7a and b). We confirmed specificity for the Gal-lectin with a polyclonal anti-Gal-lectin antibody that has been previously shown to effectively inhibit cell attachment and soluble Gal-lectin-induced responses.12 As we anticipated, treatment of ameba with the polyclonal anti-Gal-lectin antibody dose-dependently inhibited ameba-induced caspase-1 and IL-1β processing and release (Figures 7c and d). Together, these data demonstrate surface-bound Gal-lectin is required for activating the inflammasome.

Surface-bound Entamoeba histolytica (Eh) Gal-lectin is required to activate the inflammasome. (a–d) Galactose (55 mM) was used to competitively inhibit Gal-lectin binding or glucose (55 mM) as an osmotic control was added to PMA-differentiated THP-1 macrophages or lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BDMs) and exposed to 1.5 × 103 Eh for 30 min. Secretion of processed caspase-1 (a, d) and interleukin (IL)-1β (d) was detected in cell supernatants (SN) by immunoblot. Secretion of IL-1β (b) and IL-18 (c) were measured by enzyme-linked immunosorbent assay (ELISA). ***P<0.005 vs. negative control. n=4.

Surface-bound Entamoeba histolytica (Eh) Gal-lectin is required to activate the inflammasome. (a, b) Ameba were incubated with increasing concentrations of purified human colonic mucin for 10 min at 4 °C before incubation with PMA-differentiated THP-1 macrophages for 30 min. (c, d) Ameba were treated with polyclonal anti-Gal-lectin sera or normal rabbit sera at 1:100 with the indicated dilution for 10 min at 4 °C before incubation with PMA-differentiated THP-1 macrophages for 30 min. Secretion of processed caspase-1 (a, c) was detected in cell supernatants (SN) by immunoblot. Secretion of interleukin (IL)-1β (b, d) was measured by enzyme-linked immunosorbent assay (ELISA). ***P<0.005 vs. negative control. n=4. LPS, lipopolysaccharide.
Adherence epitopes of Gal-lectin modulate inflammasome activation
Next, we addressed which adherence-mediating epitopes of the Gal-lectin regulated inflammasome activation. The Gal-lectin is a single-pass transmembrane protein, composed of a large extracellular domain and a short C-terminal cytoplasmic domain. Three main structural motifs comprise the extracellular domain, an N-terminal cysteine-poor domain (residues 1–436), a pseudo-repeat domain (residues 436–624), and a cysteine-rich domain (residues 624–1,053).22 Antibodies directed at the cysteine-rich region of the Gal-lectin can either inhibit or enhance the activity of the Gal-lectin (Figure 8a).19, 20 These effects are independent of the Fc region and antibody cross-linking, as univalent Fab fragments exert identical enhancing or inhibitory activity.19 We therefore tested inflammasome activation in the presence of three adherence-inhibiting monoclonal antibodies (mAbs 1G7, 8C12, and H85) and one adherence-enhancing monoclonal antibody (mAb 3F4) that recognize non-overlapping epitopes of the Gal-lectin cysteine-rich domain (Figure 8a).19, 20 As predicted, the adherence-blocking monoclonal antibodies impaired inflammasome activation in response to ameba, as shown by reduced secretion of processed caspase-1 and IL-1β (Figures 8b and c). Consistent with previous studies that showed mAb 1G7 most effectively blocks cell adherence and soluble Gal-lectin-induced transcriptional upregulation of tumor necrosis factor-α (TNF-α) in macrophages,12, 19 we found that addition of mAb 1G7 caused the greatest inhibition of caspase-1 and IL-1β processing and release in response to ameba (Figures 8b and c). Secretion of IL-18, IL-1α, FGF-2, and MDC were also abolished by mAb 1G7 (Figure 8d, Supplementary Figures S5f–h). Identical results were obtained in BDMs, where mAb 1G7 completely inhibited release of the active caspase-1 p10 subunit and active 17 kDa IL-1β in response to ameba (Figure 8e). Thus, the epitope of Gal-lectin that mediates inflammasome activation in macrophages is highly conserved.

Adherence-mediating epitopes of Entamoeba histolytica (Eh) Gal-lectin regulate inflammasome activation. (a) Schematic diagram of the 170 kDa Gal-lectin extracellular domain and the regions recognized by the anti-Gal-lectin mAb 1G7, 3F4, 8C12, and H85. (b-e) Ameba were treated with monoclonal antibodies (mAbs) against the Gal-lectin at 1:100 dilution for 10 min at 4 °C before incubation with PMA-differentiated THP-1 macrophages or lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BDMs) for 30 min. (b, e) Secretion of processed caspase-1 and IL-1β was detected in cell supernatants (SN) by immunoblot. Secretion of IL-1β (c) and IL-18 (d) were measured by ELISA. ***P<0.005 vs. negative control. n=4.
We used the caspase-1-FLICA to visualize the effects of Gal-lectin inhibition on a single-cell level. As expected, we rarely observed ameba attached to macrophages after exposure to mAb 1G7 and correspondingly the caspase-1-FLICA labeled only a minor proportion of macrophages exposed to ameba treated with mAb 1G7 (Figure 9). Occasionally, ameba appeared to be in close proximity to macrophages, but there was no active caspase-1 (Figure 9). This indicates that Eh in the absence of Gal-lectin binding is not sufficient to evoke inflammasome activation, suggesting Gal-lectin may deliver a specific ‘adherence’ signal to trigger the inflammasome. We observed modest inhibition of caspase-1 and IL-1β activation by mAb 8C12 and mAb H85, which is consistent with previous studies12, 19, 20 showing they are less effective than mAb 1G7 at inhibiting Gal-lectin binding to cells and soluble Gal-lectin-induced transcriptional upregulation of TNF-α in macrophages (Figures 8b and c). However, the functions of these epitopes are not entirely conserved, as in some cell types mAb 8C12 blocks adherence as effectively as 1G7, whereas mAb H85 has no effect on Gal-lectin binding to intestinal mucin.19, 20 This probably reflects different binding mechanisms of Gal-lectin to different host receptors. Interestingly, however, when ameba were exposed to mAb 3F4, which enhances Gal-lectin-binding activity, we observed a significant increase in the processing and secretion of caspase-1 and IL-1β (Figures 8b and c). Addition of an immunoglobulin G isotype control had no effect on ameba-induced caspase-1 and IL-1β activation (Figures 8b–e). Thus, these data show and corroborate our previous results that attachment via surface-bound Gal-lectin was required for activating the inflammasome.

Adherence-mediating epitopes of Entamoeba histolytica (Eh) Gal-lectin regulate inflammasome activation. Amebas were treated with Gal-lectin inhibitory mAb 1G7 at 1:100 dilutions for 10 min at 4 °C before incubation with PMA-differentiated THP-1 macrophages where active caspase-1 is labeled by YVAD-FLICA. Mammalian nuclei are labeled with 4,6-diamidino-2-phenylindole (DAPI; ameba nuclei are not stained by DAPI). Confocal images showing Eh in contact (white arrow) with macrophages activated caspase-1 (center panel) whereas no Eh adhered to macrophages following treatment with mAb 1G7. The Eh+1G7 panel shows an Eh in close proximity with a macrophage (Mφ) in the absence of caspase-1 activation. Scale bar=20 μm.
Soluble Gal-lectin is immunostimulatory but does not induce inflammasome activation
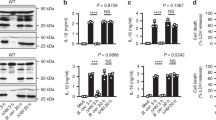
Gal-lectin is continually shed and replenished on the ameba membrane, and is a constituent of viable ameba secreted components. As our results revealed that inflammasome activation was uniquely contact dependent, they predicted that Gal-lectin in its soluble form would not trigger the inflammasome. Thus, to determine whether Gal-lectin could directly induce inflammasome activation we exposed differentiated THP-1 macrophages and BDMs to purified native soluble Gal-lectin. As predicted, no concentration tested activated the inflammasome in either cell type (Figures 10a–c). Even after several hours, soluble Gal-lectin did not induce release of IL-1β, IL-1α, FGF-2, MDC, and IP-10 as measured by multiplex cytokine array ( Supplementary Figures S6a–e ). Moreover, addition of soluble Gal-lectin dose-dependently attenuated release of caspase-1 and IL-1β in response to ameba, demonstrating soluble Gal-lectin blocks the receptors on macrophages that mediate contact-dependent inflammasome activation (Figures 10d and e). The soluble Gal-lectin was immunostimulatory, however, as it dose-dependently induced TNF-α release, which is an inflammasome-independent cytokine and a known Gal-lectin-induced response (Figure 10f).12, 13 TNF-α release was specific as it was inhibited by mAb 1G7 but not the isotype control (Figure 10f). As transcriptional priming is a critical step for activating the inflammasome in naive macrophages, we tested whether soluble Gal-lectin could induce expression of inflammasome components that are known to require transcriptional upregulation; namely pro-IL-1β and NLRP3, which are induced via nuclear factor-κB.23 Soluble Gal-lectin upregulated the expression of pro-IL-1β and NLRP3 as efficiently as LPS and was sufficient to fulfill the priming requirement for NLRP3 activation in response to nigericin (Figures 10g and h).

Soluble native Entamoeba histolytica (Eh) Gal-lectin is immunostimulatory but does not activate the inflammasome. (a, b) PMA-differentiated THP-1 macrophages or lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BDMs) were exposed to live Eh or soluble Gal-lectin (1 μg ml−1) for 30 min. (d, e) Increasing concentrations of soluble Gal-lectin were added to PMA-differentiated THP-1 macrophages and stimulated with 1.5 × 103 live Eh for 30 min. Secretion of processed caspase-1 (a, c, d) and interleukin (IL)-1β (c) was detected in cell supernatants (SN) by immunoblot and IL-1β secretion was measured by enzyme-linked immunosorbent assay (ELISA; b, e). (f) Tumor necrosis factor α (TNF-α) release by PMA-differentiated THP-1 macrophages exposed to the indicated number of live ameba or soluble Gal-lectin for 3 h measured by ELISA. Specificity for the Gal-lectin was shown by pre-treating the Gal-lectin with adherence inhibitory mAb 1G7 or immunoglobulin G (IgG) control (1:100 dilution) for 10 min at 4 °C before incubation with macrophages. (g) Immunoblot showing the expression of pro-IL-1β, NLRP3, and pro-caspase-1 in the lysates of BDM macrophages primed with LPS (1 μg ml−1) or Gal-lectin (1 μg ml−1) for 4 h. (h) BDM were primed with Gal-lectin (1 μg ml−1) for 4 h and nigericin (10 μM) was added in the final 30 min. Active caspase-1 and IL-1β were detected in cell SNs by immunoblot. **P<0.01, ***P<0.005 vs. negative control. n=4. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; LYS, lysates.
Discussion
Eh exists entirely as an extracellular pathogen and inflicts damage primarily through adherence to host cells, which enables the parasite to kill or ingest its target.2 Adherence is therefore a central feature of Eh pathogenicity. The PRRs that respond to ameba adherence are not known. In this study, we showed the inflammasome uniquely senses contact with intact live ameba, as neither secreted components of viable ameba nor whole lysed ameba trigger inflammasome activation. Based on these intriguing results, we propose the inflammasome distinguishes between the physical forms of ameba, that is, whether the cell has encountered the surface of intact ameba or components that have been released by distant ameba or debris of dead ameba. This mode of inflammasome activation suggests that during amebic invasion the inflammasome is a pathogenicity sensor when Eh poses the highest level of danger to the host and predicts that it coordinates host defenses that eliminate intact ameba.
What are the consequences of inflammasome activation for host defense? We observed rapid and robust secretion of IL-1β, as well as IL-18, IL-1α, FGF-2, IP-10, and MDC that would shape the magnitude of the local inflammatory response. Eh-induced inflammasome activation does not appear to trigger cell death, which often occurs following caspase-1 activation. As activated macrophages are directly amebicidal,24 inflammasome-induced death would likely hinder elimination of invasive ameba and be detrimental to host defense. Future studies are required to determine whether the inflammasome regulates direct amebicidal responses in macrophages.
The other major implication of our study is that in different contexts Gal-lectin has a very different role in alerting the host to infection. We showed that when ameba are not directly present, soluble Gal-lectin stimulates TNF-α release and transcriptional activation of the inflammasome. On the other hand, when intact ameba ligates host cells Gal-lectin has an essential role in activating the inflammasome. How does soluble Gal-lectin and surface-bound Gal-lectin stimulate distinct responses? We envision several possibilities. First another PRR may mediate soluble Gal-lectin induced responses. We have previously reported that soluble Gal-lectin activates the nuclear factor-κB p65 subunit similar to LPS.14, 25 In this study, we showed that Gal-lectin is equivalent to LPS in upregulating pro-IL-1β and NLRP3 expression, and fulfills the priming requirements for NLRP3 activation, which all require nuclear factor-κB activation. Gal-lectin may thus stimulate a Toll-like receptor, which respond to soluble ligands.
In the context of an intact ameba, surface-bound Gal-lectin may simply tether Eh to host cells and deliver another parasite molecule into close proximity with the macrophage that activates the inflammasome. Alternately, surface-bound Gal-lectin might actively participate in triggering the inflammasome. This could occur by cross-linking a host membrane receptor, inducing a higher level of activation than soluble Gal-lectin, to supply additional signaling leading to inflammasome activation. Previous studies have reported contact by intact ameba results in massive reorganization of the host cell cytoskeleton and focal adhesions.26, 27 During contact, Gal-lectin-induced cross-linking of a host receptors could disrupt movement of the plasma membrane or plasma membrane proteins, and this event could be sensed by the inflammasome. Future studies are required to determine the features of contact that underlie the mechanism of inflammasome activation.
Methods
Cultivation, harvesting of E. histolytica , and amebic liver abscess in gerbils. E. histolytica HM-1:IMSS were grown axenically in TYI-S-3 medium with 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate at 37 °C in sealed 15 ml borosilicate glass tubes as described previously.28 To maintain virulence, trophozoites were regularly passed through gerbil livers as described.24 Ameba were harvested after 72 h of growth by centrifugation at 200 × g for 5 min at 4 °C and resuspended in serum-free RPMI. For establishing amebic liver abscess, male Mongolian gerbils from Charles River Laboratories (St Constant, Quebec, Canada) were inoculated into the left liver lobe with log-phase virulent ameba as described previously16 and animals killed at 10 days post infection. Serum was pooled from six animals and cytokines were measured using a rat multiplex cytokine array by Eve Technologies (Calgary, Alberta, Canada).
Reagents and cytokine quantification. Ultra-pure LPS, nigericin, phorbol-12-myristate-13-acetate, glucose, and galactose were from Sigma-Aldrich (St Louis, MO). Z-VAD(OMe)-FMK and Z-YVAD(OMe)-FMK were from Enzo Life Sciences (Plymouth Meeting, PA). Human IL-1β enzyme-linked immunosorbent assay (ELISA) was from R&D Systems (Minneapolis, MN), human IL-18 ELISA was from MBL International (Woburn, MA), and human TNFα ELISA was from eBiosciene (San Diego, CA). Human 41-plex cytokine arrays was done by Eve Technologies and used to measure IL-1α, FGF-2, IP-10, and MDC. The generation of mouse anti-Gal-lectin mAbs 1G7, H85, 8C12, and 3F4 have been described.19 The recognition sites of these mAbs have been mapped using successive deletion peptides of the 170 kDa Gal-lectin.20 Human immune serum from patients with amebic liver abscess was provided courtesy of J Keystone, University of Toronto. Polyclonal anti-Gal-lectin serum was prepared in rabbits inoculated with native purified Gal-lectin.12
Preparation of amebic proteins, native soluble Gal-lectin, and colonic mucin. Concentrated secreted components were collected from live ameba (2 × 107 ml−1) incubated in Hank’s balanced salt solution for 2 h at 37 °C. Fresh whole amebic lysates were prepared by freeze-thaw lysis of ameba in Hank’s balanced salts solution. Ameba membrane and cytosolic fractions were prepared by centrifuging fresh freeze-thawed lysates at 4 °C for 15 min at 14,000 × g. Supernatants and resuspended pellets were used for cytosolic components and membrane components, respectively. Purification of the native Gal-lectin by mAb affinity chromatography has been described.10 Briefly, octyl glucoside-solublized ameba was applied to a column consisting of purified anti-Gal-lectin mAbs H-86, 7F4, 5B8, 3F4, and 6D2. Human colonic mucin from LS 174T cells was prepared as described.29 In brief, lyophilized crude mucus preparations were resuspended in 0.01 Tris-HCL, 0.001% sodium azide and applied to a Sepharose 4B column. High molecular weight carbohydrate-rich mucin fractions isolated in void volume was pooled and dialyzed exhaustively against deionized water at 4 °C, lyophilized, and resuspended in water.
Human and mouse macrophage studies. THP-1 human monocytic cells (ATCC, Manassas, VA) were cultured in RPMI-1640 with 10% fetal bovine serum, 10 mM HEPES, 50 mM 2-mercaptoethanol, and antibiotics. Cells were maintained in a humidified incubator with 5% CO2. For experiments, 4 × 105 THP-1 cells per well were seeded onto 24-well plates in complete RPMI containing phorbol-12-myristate-13-acetate (50 ng ml−1) the night before experiment. For experiments, cells were stimulated in a final volume of 250 μl serum-free RPMI-1640. Wt C57BL/6 mice were from Charles River Laboratories and caspase-1−/− mice on a C57BL/6 background were obtained from Dr Yan Shi (University of Calgary). BDMs were prepared from the femurs and tibias of mice cultured for 6 days in 10% FBS-RPMI-1640 supplemented with 30% L-cell supernatant, then plated onto 24-well plates at 5 × 105 cells per well in complete RPMI-1640. On the day of experiment, BDMs were treated with 1 μg ml−1 LPS for 4 h. In the final 30 min, stimuli were added to BDM in 250 μl of serum-free RPMI. The University of Calgary Animal Care Committee approved all experiments involving animals.
Immunoblotting. THP-1 or BDM supernatants from four wells were pooled and centrifuged at 4 °C for 5 min at 2,000 × g. Pelleted debris was discarded and supernatants were concentrated by trichloro acetic acid precipitation. For immunoblotting blotting, precipitated supernatants were resuspended in 50 μl Laemmli buffer, boiled for 5 min and equal volumes were resolved on 12.5% polyacrylamide gels and transferred to nitrocellulose. For cell lysates, plates were washed in cold phosphate-buffered saline before adding lysis buffer (NaCL, Tris (pH 8), SDS, Triton 100-X, EDTA, phenylmethylsulfonyl fluoride, E-64, leupeptin, aprotonin, supplemented with protease inhibitor cocktail (Sigma-Aldrich)) and centrifuged at 4 °C for 15 min at 14,000 g. Equal amounts of proteins boiled for 5 min in Laemmli buffer were resolved on 7.5–12.5% polyacrylamide gels and transferred to nitrocellulose. Membranes were blocked in 5% skim milk, incubated overnight at 4 °C in primary Abs, and visualized with secondary horseradish peroxidase-conjugated Abs. Supernatants were detected with SuperSignal Chemiluminescence Reagents (Pierce, Rockford, IL) and lysates with ChemiLucent ECL detection (EMD Millipore, Billerica, MA). Primary Abs were anti-IL-1β cleaved human (2021, Cell Signaling, Beverly, MA), anti-caspase-1 human (2225, Cell Signaling), anti-caspase-1 human (622, Santa Cruz Technologies, Santa Cruz, CA), anti-IL-1β mouse (AF401, R&D Systems), anti-caspase-1 mouse (514, Santa Cruz Technologies), anti-NLRP3 (AG-20B-0014, Adipogen, San Diego, CA), and anti-glyceraldehyde 3-phosphate dehydrogenase (Jackson ImmunoResearch, West Grove, PA).
Confocal microscopy. THP-1 cells were seeded on polylysine-coated glass coverslips and stimulated with ameba or LPS (25 ng ml−1) and nigericin (10 μM). Cells were washed in cold phosphate-buffered saline and fixed in cold acetone for 5 min. YVAD-FLICA (FLICA 660-Caspase-1 reagent, Immunochemistry Technologies, Bloomington, MN) at 1:70 dilution was added for 1 h at room temperature. Cells were washed twice in phosphate-buffered saline-Tween (0.1%) for 5 min and mammalian nuclei were stained with 4,6-diamidino-2-phenylindole for 20 min, followed by two washes in phosphate-buffered saline-Tween (0.1%) for 5 min. Cells were mounted onto slides and imaged immediately as YVAD-FLICA staining dissipates quickly after fixation. Cells were analyzed on an Olympus IX8-1 FV1000 Laser Scanning Confocal microscope (Tokyo, Japan) with a 60 × objective.
Statistics. All experiments shown are representative of three independent experiments unless otherwise indicated. GraphPad Prism4 (San Diego, CA) was used for statistical analysis. Treatment groups were compared using the paired Student’s t-test. Statistical significance was assumed at P<0.05. Results are displayed as mean+s.e.m.
References
Stanley, S.L. Amoebiasis. Lancet 361, 1025–1034 (2003).
Mortimer, L. & Chadee, K. The immunopathogenesis of Entamoeba histolytica. Exp. Parasitol. 126, 366–380 (2010).
Prathap, K. & Gilman, R. The histopathology of acute intestinal amebiasis. Am. J. Pathol. 60, 229–246 (1970).
Chadee, K. & Meerovitch, E. Entamoeba histolytica: early progressive pathology in the cecum of the gerbil (Meriones unguiculatus). Am. J. Trop. Med. Hyg. 34, 283–291 (1985).
Seydel, K.B., Li, E., Swanson, P. & Stanley, S.L. Human intestinal epithelial cells produce pro-inflammatory cytokines in response to infection in a SCID mouse-human intestinal xenograft model of amebiasis. Infect. Immun 65, 1631–1639 (1997).
Seydel, K.B., Li, E., Zhang, Z. & Stanley, S.L. Epithelial cell-initiated inflammation plays a crucial role in early tissue damage in amebic infection of human intestine. Gastroenterology 115, 1446–1453 (1998).
Kissoon-Singh, V., Moreau, F., Trusevych, E. & Chadee, K. Entamoeba histolytica exacerbates epithelial tight junction permeability and pro-inflammatory responses in Muc2−/− mice. Am. J. Pathol 182, 852–865 (2013).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Keller, M., Rüegg, A., Werner, S. & Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831 (2008).
Petri, W.A., Smith, R.D., Schlesinger, P.H., Murphy, C.F. & Ravdin, J.I. Isolation of the galactose-binding lectin that mediates the in vitro adherence of Entamoeba histolytica. J. Clin. Invest. 80, 1238–1244 (1987).
Chadee, K., Petri, W.A., Innes, D.J. & Ravdin, J.I. Rat and human colonic mucins bind to and inhibit adherence lectin of Entamoeba histolytica. J. Clin. Invest. 80, 1245–1254 (1987).
Séguin, R., Mann, B.J., Keller, K. & Chadee, K. Identification of the galactose-adherence lectin epitopes of Entamoeba histolytica that stimulate tumor necrosis factor-α production by macrophages. Proc. Natl. Acad. Sci. USA 92, 12175–12179 (1995).
Séguin, R., Mann, B.J., Keller, K. & Chadee, K. The tumor necrosis factor alpha-stimulating region of galactose-inhibitable lectin of Entamoeba histolytica activates gamma interferon-primed macrophages for amebicidal activity mediated by nitric oxide. Infect. Imm 65, 2522–2527 (1997).
Ivory, C. & Chadee, K. Activation of dendritic cells by the Gal-lectin of Entamoeba histolytica drives Th1 responses in vitro and in vivo. Eur. J. Immunol 37, 385–394 (2007).
Houpt, E. et al. Prevention of intestinal amebiasis by vaccination with the Entamoeba histolytica Gal/GalNac lectin. Vaccine 22, 611–617 (2004).
Haque, R., Ali, I.M., Sack, R.B., Farr, B.M., Ramakrishnam, G. & Petri, W.A. Amebiasis and mucosal IgA antibody against the Entamoeba histolytica adherence lectin in Bangladeshi children. J. Infect. Dis. 183, 1787–1793 (2001).
Chadee, K. & Meerovitch, E. The pathogenesis of experimentally induced amebic liver abscess in the gerbil (Meriones unguiculatus). Am. J. Pathol 117, 71–80 (1984).
Broz, P., von Molyke, J., Jones, J.W., Vance, R.E. & Monack, D. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Petri, W.A. et al. Monoclonal antibodies directed against the galactose-binding lectin of Entamoeba histolytica enhance adherence. J. Immunol 144, 4803–4809 (1990).
Mann, B.J., Chung, C.Y., Dodson, J.M., Ashley, L., Braga, L.L. & Snodgrass, T.L. Neutralizing monoclonal antibody epitopes of the Entamoeba histolytica galactose adhesin map to the cysteine-rich extracellular domain of the 170-kilodalton subunit. Infect. Immun 61, 1772–1778 (1993).
Ravdin, J.I., Petri, W.A., Murphy, C.F. & Smith, R.D. Production of mouse monoclonal antibodies which inhibit in vitro adherence of Entamoeba histolytica. Infect. Immun. 53, 1–5 (1986).
Lotter, H., Zhang, T., Seydel, K.B., Stanley, S.L. & Tannich, E. Identification of an epitope on the Entamoeba histolytica 170-kD lectin conferring antibody-mediated protection against invasive amebiasis. J. Exp. Med. 185, 1793–1801 (1997).
Bauernfeind, F.G. et al. NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol 183, 787–791 (2009).
Denis, M. & Chadee, K. Cytokine activation of murine macrophages for in vitro killing of Entamoeba histolytica trophozoites. Infect. Immun 57, 1750–1756 (1989).
Kammanadiminti, S., Mann, B.J., Dutil, L. & Chadee, K. Regulation of Toll-like receptor-2 expression by the Gal-lectin of Entamoeba histolytica. FASEB J 18, 155–157 (2004).
Faust, D.M., Markiewicz, J.M., Danckaert, A., Soubigou, G. & Guillen, N. Human liver sinusoidal endothelial cells respond to interaction with Entamoeba histolytica by changes in morphology, integrin signalling and cell death. Cell. Microbiol. 13, 1091–1106 (2011).
Lee, Y.A., Kim, K.A. & Shin, M.H. Calpain mediates degradation of cytoskeletal proteins during Jurkat T-cell death induced by Entamoeba histolytica. Parasite. Immunol 33, 349–356 (2011).
Diamond, L.S., Harlow, D.R. & Cunnick, C.C. A new medium for the axenic cultivation of Entamoeba histolytica. Trans. R. Soc. Trop. Med. Hyg. 72, 431–432 (1978).
Belly, A., Keller, K., Grove, J. & Chadee, K. Interaction of LS 174 T human colon cancer cell mucins with Entamoeba histolytica: an in vitro model for colonic disease. Gastroenterology 111, 1484–1492 (1996).
Acknowledgements
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to KC. LM is supported by Achievers in Medicine Award and NSERC and Alberta Innovates Health Solution graduate scholarships. SC is supported by an NSERC CREATE Collaborative Research and Training Experience Grant. We thank Dr Pina Colarruso for technical assistance from the Live Cell Imaging Core for confocal studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article
Cite this article
Mortimer, L., Moreau, F., Cornick, S. et al. Gal-lectin-dependent contact activates the inflammasome by invasive Entamoeba histolytica. Mucosal Immunol 7, 829–841 (2014). https://doi.org/10.1038/mi.2013.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.100
This article is cited by
-
Entamoeba histolytica exploits the autophagy pathway in macrophages to trigger inflammation in disease pathogenesis
Mucosal Immunology (2021)
-
Entamoeba histolytica stimulates the alarmin molecule HMGB1 from macrophages to amplify innate host defenses
Mucosal Immunology (2020)
-
Entamoeba histolytica-induced IL-1β secretion is dependent on caspase-4 and gasdermin D
Mucosal Immunology (2019)
-
Inflammasomes and intestinal inflammation
Mucosal Immunology (2017)
-
Immune responses against protozoan parasites: a focus on the emerging role of Nod-like receptors
Cellular and Molecular Life Sciences (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}