
Abstract
The αE integrin chain CD103 identifies a subset of migratory dendritic cells (DCs) in the gut, lung, and skin. To gain further understanding of the function of CD103+ DCs in regulating adaptive immunity in vivo, we coupled ovalbumin (OVA) to the CD103 antibody M290 (M290.OVA). Intraperitoneal injection of M290.OVA induced OVA-specific CD8+ and CD4+ T-cell proliferation in lymph nodes (LNs) of wild-type but not CD103−/− mice, or in mice depleted of CD11c+ cells. In the absence of maturation stimuli, systemic antigen targeting to CD103+ DCs led to tolerance of CD8+ T cells, whereas coadministration of adjuvant induced cytotoxic T-lymphocyte (CTL) immunity and antibody production. Mucosal intratracheal application of M290.OVA also induced T-cell proliferation in mediastinal LNs, yet the functional outcome was tolerance that inhibited subsequent development of allergic airway inflammation and immunoglobulin E (IgE) responses to inhaled OVA. These findings identify antigen targeting to CD103+ DCs as a potential strategy to regulate immune responses in nonlymphoid mucosal tissues.
Similar content being viewed by others


Introduction
Conventional dendritic cells (DCs) represent a heterogeneous group of antigen-presenting cells located throughout lymphoid and extralymphoid tissues of the body that together play a fundamental role in the initiation and diversification of adaptive immune responses. Although lymphoid-resident DC subsets have been the subject of intensive investigation and display distinct functional properties,1, 2, 3 the functional specialization of peripheral tissue-derived migratory DC subsets is less well defined. Recent studies by us, and others, have identified a major subset of migratory DCs in the intestine, lung, and dermis that expresses the αE integrin chain CD103.4, 5, 6, 7, 8, 9, 10, 11
Several functions have been ascribed to peripheral tissue CD103+ DCs including a role in cross-presenting viral and self-antigens in lung and skin-draining LNs,8, 12, 13, 14 cross-presenting innocuous inhaled antigen in bronchial LNs,15 and initiating CD4+ and CD8+ T-cell responses to soluble intestinal antigens in mesenteric LNs (MLNs).5, 16 In addition, murine and human small intestinal lamina propria (SI-LP) and MLN CD103+ DCs display an enhanced ability to induce the gut-homing receptors CCR9 and α4β7 on responding T cells and promote naive CD4+ T-cell differentiation to regulatory T cells (Tregs) in vitro.4, 5, 17, 18, 19, 20 Nevertheless, many of these studies have relied on assessing the function of CD103+ DCs ex vivo, and the in vivo antigen-presenting capacity of these cells remains unclear.
In this study we demonstrate that the CD103 antibody M290 can be utilized to selectively target antigen to murine CD103+ DCs and utilize this strategy to assess the functional capacity of this major peripheral tissue migratory DC subset in vivo.
Results
M290 is internalized by CD103+ MLN DCs and associates with early and late endosomes
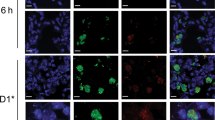
The rat IgG2a CD103 antibody M290 is a nondepleting antibody,21 and in vivo administration of M290 coupled to the toxin saporin (M290-SAP) leads to the selective depletion of CD103+ cells including CD103-expressing CD8+ T cells, DCs, and Tregs.21 To assess whether CD103 could be utilized as a target to deliver antigen to CD103+ DCs, MLN DCs from wild-type and CD103−/− mice were incubated with M290 or isotype control (GLIII/10) antibody. As expected, M290 but not GLIII/10 bound a subset of MLN DCs from wild-type but not CD103−/− mice (Supplementary Figure S1a online). Similar to antibodies targeting the C-type lectins CD205 and mDCIR1,22, 23 following 30 min of incubation at 37 °C, M290 was taken up by MLN DCs and colocalized with early endosome antigen-1 (EEA-1)+ early endosomes (Figure 1a and Supplementary Video S1 online, data not shown), and at 60 min was occasionally observed to be associated with lysosomal-associated membrane protein-1 (LAMP-1)+ late endosomes/lysosomes (Figure 1b and Supplementary Video S2 online). Low cell viability after in vitro culture precluded assessment at later time points. Colocalization of M290 with EEA-1+ or LAMP-1+ vesicles was not observed when MLN DCs were maintained on ice (data not shown).

In vivo M290.OVA-targeted CD103+ dendritic cells (DCs) induce OT-1 proliferation ex vivo. (a, b) Magnetic-activated cell sorting (MACS)-purified CD11c+ mesenteric lymph node (MLN) cells were incubated with M290 or GLIII/10, washed, and incubated at 37 °C for the indicated time. Cells were then fixed, permeabilized, and costained for (a) the early endosomal marker EEA-1 (early endosome antigen-1) or (b) EEA-1 and the late endosomal/lysosomal marker, lysosomal-associated membrane protein-1 (LAMP-1). Results are representative cells from one experiment of three performed. (c) C57BL/6 mice were injected intraperitoneally (IP) with M290.OVA (10 μg, clear, solid line) or GLIII/10.OVA (10 μg, filled). Mice were killed after 17 h, and MLN cells were stained with PE-M290 (upper panel) or anti-rat IgG2a (lower panel). Plots are gated on MHCII+CD11c+ cells. Untreated mice (clear, dotted line). Results from one representative experiment of three performed is shown. (d) C57BL/6 mice were injected IP with M290.OVA (10 μg) or GLIII/10.OVA (20 μg). At 17 h after immunization, CD103+ and CD103− MHCII+ CD11c+ DCs and CD103+ and CD103− non-DCs were sorted from the MLNs and their ability to induce OT-I proliferation assessed by thymidine incorporation. Proliferation is presented as the percentage of [3H]-thymidine incorporation observed in T cells cultured with CD103+ DCs from M290.OVA-treated mice (mean c.p.m. 5,045) and is presented in this way to normalize between individual experiments. Results are the mean of 3–5 independent experiments using pooled cells from 7 mice per condition and experiment. ***P<0.001.
To confirm that M290 targets CD103+ DCs in vivo, fluorescently labeled M290 or GLIII/10 were injected intraperitoneally (IP) into C57BL/6 mice. After 17 h, animals were killed and tissues were analyzed by flow cytometry (Supplementary Figure S1b online). M290 was detected on CD11c+MHCII+ cells in MLN and SI-LP but less in the spleen, consistent with previous observations that CD103+ DCs are rare in the spleen.4, 5 A similar proportion of CD103+ DCs was observed in uninjected mice after staining for CD103 in vitro (data not shown), demonstrating efficient in vivo targeting of DCs in these tissues. Analysis of MLN tissue sections demonstrated colocalization of M290 with CD11c+ cells (Supplementary Figure S1c online). M290 also bound to CD8+ cells, consistent with expression of CD103 on naive and gut-resident CD8+ T cells,24, 25 but not B220+ cells (Supplementary Figure S1b online). Together, these results demonstrate that M290 is internalized by CD103+ DCs and can be used to target CD103+ DCs in vivo.
M290.OVA conjugates induce OT-I cell proliferation in vitro and ex vivo
To assess the in vivo antigen-presenting capacity of CD103+ DCs, M290 and GLIII/10 were chemically coupled to the model antigen ovalbumin (OVA) (Supplementary Figure S2a online). At the concentrations tested, M290.OVA- but not GLIII/10.OVA-pulsed MLN DCs induced dose-dependent OT-I proliferation in vitro (Supplementary Figure S2b online). This was because of specific targeting and presentation by CD103+ DCs as M290.OVA-pulsed CD103− MLN DCs failed to induce OT-I proliferation (Supplementary Figure S2c online).
To determine whether M290.OVA efficiently targets CD103+ DCs in vivo, mice were injected with antibody/OVA conjugates IP and animals were killed 17 h later (Figure 1c). MLN DCs from mice injected with M290.OVA stained with anti-rat secondary antibody but failed to bind directly conjugated M290. In contrast, MLN DCs from mice that had received GLIII/10.OVA stained with fluorescently labeled M290 but not with anti-rat secondary antibody (Figure 1c). Thus, CD103-expressing DCs were selectively and efficiently targeted by M290.OVA, whereas GLIII/10.OVA failed to bind DCs in vivo.
To assess whether DCs from mice receiving antibody conjugates in vivo induced OT-I priming ex vivo, antibody/OVA complexes were injected IP and 17 h later CD103+ and CD103− DCs and non-DCs were sorted from the MLNs and co-cultured with OT-I cells (Figure 1d). Only CD103+ MLN DCs from animals receiving M290.OVA induced efficient OT-I proliferation, whereas cells from mice receiving GLIII/10.OVA induced very weak or no proliferation. Collectively, these results demonstrate that the M290.OVA conjugates target CD103+ DCs in vivo and that these cells can process such conjugates for induction of CD8+ T-cell responses.
Systemic administration of M290.OVA induces OT-I and OT-II proliferation in vivo
To determine whether M290.OVA could induce T-cell responses in vivo, carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-I.CD45.1 and OT-II.CD45.1 cells were transferred into C57BL/6 mice and recipient animals were injected with antibody/OVA conjugates together with lipopolysaccharide (LPS) 1 day later. The amount of each antibody conjugate utilized for injection was normalized to the levels of OVA bound to each antibody as determined by enzyme-linked immunosorbent assay and are depicted in the figure legends. After 3 days, MLNs were isolated from recipient animals and OT-I and OT-II proliferation assessed by flow cytometry. M290.OVA but not GLIII/10.OVA and LPS induced both OT-I and OT-II proliferation in vivo (Figure 2a,b). This response was dependent on specific targeting of the conjugate to CD103+ antigen-presenting cells as OT-I and OT-II cells failed to proliferate in CD103−/− mice (Figure 2c,d). The lack of proliferation in CD103−/− mice was not because of a general priming defect in these animals as administration of OVA (0.5 mg) IP induced similar T-cell responses in CD103−/− and wild-type mice (Supplementary Figure S3a and b online). Importantly, OT-I and OT-II cells did not proliferate in mice injected with OVA (1 μg) given together with unconjugated M290 and LPS, demonstrating a requirement for selective targeting of OVA to CD103+ cells (Supplementary Figure S4a and b online). Finally, to confirm a role for CD103+ DCs in T-cell priming, diphtheria toxin (DTx)-treated or untreated CD11c.DTR mice were injected with CFSE-labeled OT-I/OT-II cells and M290.OVA and LPS. M290.OVA and LPS induced OT-I and OT-II proliferation in the MLNs and mediastinal LNs in untreated but not DTx-treated CD11c.DTR mice. Efficient T-cell priming was also observed in the spleen of untreated but not DTx-treated mice, indicating that the few CD103+ DCs present in the spleen also function in vivo as efficient antigen-presenting cells (Supplementary Figure S5 online). Together, these results provide strong evidence that CD103+ DCs induce both CD8+ and CD4+ T-cell responses in vivo.

M290.OVA induces OT-I and OT-II T cell proliferation in vivo. (a–d) C57BL/6 or (c, d) CD103−/− mice were injected intravenously (IV) with carboxyfluorescein succinimidyl ester (CFSE)-labeled CD45.1+ OT-I and OT-II cells. PBS, phosphate-buffered saline. After 24 h, mice were immunized intraperitoneally (IP) with (a–d) M290.OVA (1 μg) and lipopolysaccharide (LPS; 50 μg) or (a, b) GLIII/10.OVA (2 μg) and LPS (50 μg). At 3 days after immunization, mesenteric lymph nodes (MLNs) were collected and the proliferation (a, c) and cell number (b, d) of OT-I/OT-II cells assessed by flow cytometry. (a, c) Representative flow cytometry plots from mice receiving (a) M290.OVA (clear) or GLIII/10.OVA (shaded) or (c) M290.OVA in wild-type (wt) mice (clear) or CD103−/− mice (shaded). (b, d) Mean (s.e.m.) from one representative experiment of three performed with (b) 4 and (d) 3 mice per group. *P<0.05; **P<0.01; ***P<0.001.
Systemic administration of M290.OVA generates gut-homing T cells in vivo but fails to induce efficient FoxP3+ Treg differentiation
SI-LP and MLN CD103+ DCs display an enhanced ability to induce the gut-homing receptors CCR9 and α4β7 on responding T cells in vitro.4, 5, 16, 17, 26 To assess the ability of CD103+ MLN DCs to induce gut-homing receptors on T cells in vivo, OT-I and OT-II cell recipients were injected IP with M290.OVA and LPS. After 3 days, the majority of MLN-primed OT-I cells expressed CCR9 and α4β7 (Figure 3a) and OT-I cells readily migrated to the small intestinal epithelium (Figure 3b). Although OT-II cells were also induced to express α4β7, CCR9 expression was restricted to cells that had undergone few divisions and was then lost (Figure 3c). Similar expression of gut-homing receptors was observed when M290.OVA was injected alone, or when mice were injected with FTY720 1 day after immunization to prevent effector T-cell egress (data not shown). Together, these results suggest that CD103+ MLN DCs are more efficient at inducing gut-homing receptors on CD8+ T cells than CD4+ T cells in vivo.

CD103+ mesenteric lymph node (MLN) dendritic cells (DCs) generate gut-homing T cells in vivo but are poor at inducing FoxP3 (forkhead box P3)+ regulatory T cell (Treg) differentiation. (a) Representative flow cytometry plots of gut-homing receptor expression on OT-I cells in the MLNs 3 days after immunization with M290.OVA (1 μg) and lipopolysaccharide (LPS; 50 μg). Numbers are mean (s.e.m.) positive cells among dividing cells from 10 mice in 4 separate experiments. CFSE, carboxyfluorescein succinimidyl ester. (b) C57BL/6 or CD103−/− mice were immunized with M290.OVA (1 μg) or GLIII/10.OVA (2 μg) with LPS and their migration to the small intestinal epithelium determined by flow cytometry. Results are the mean (s.e.m.) from one representative experiment of four, with three mice per group. IEL, intraepithelial lymphocyte; PBS, phosphate-buffered saline; wt, wild type. (c) Representative FACS plots of gut-homing receptor expression on OT-II cells in the MLNs 3 days after immunization with M290.OVA (1 μg) and LPS. Numbers are the mean (s.e.m.) positive cells among dividing cells from 12 mice in 4 separate experiments. (d, e) Representative flow cytometry plots of FoxP3 expression on OT-II cells in the MLNs 3 days after intraperitoneal (IP) immunization with (d) M290.OVA (1 μg) and LPS (d, e) M290.OVA (5 μg) or (e) orally with ovalbumin (OVA, 5 mg). (e) At 1 day after immunization, some of the groups were treated with FTY720 (FTY, 20 μg IP). Numbers are the mean (s.e.m.) positive cells among dividing cells from seven mice in three separate experiments. **P< 0.01; ***P< 0.001.
Recent studies have suggested that MLNs are a preferential site of peripheral FoxP3 (forkhead box P3)+ Treg conversion, although the observed proportion of converted cells generally appears low.16, 17 Given the enhanced ability of SI-LP and MLN CD103+ DCs to promote induced Treg differentiation in vitro,16, 17 we hypothesized that selective antigen presentation by CD103+ MLN DCs in vivo may dramatically enhance induced Treg conversion in the MLNs. To assess this, recipients of naive OT-II cells were immunized with M290.OVA with/without LPS IP, and FoxP3+ expression in responding cells was determined 3 days later. Under these conditions, 3–6% of OT-II cells expressed Foxp3+ (Figure 3d). Similar percentages of FoxP3+ Tregs were observed in mice that were injected with FTY720 at 1 day after immunization, to prevent lymphocyte egress (Figure 3e). Notably, a similar degree of FoxP3+ Treg generation was observed after oral OVA administration (Figure 3e), a process that requires DC-mediated antigen transport to draining MLNs.27, 28
Systemic administration of M290.OVA induces tolerance or immunity depending on the instruction of DC maturation
We next addressed the ability of M290.OVA to induce functional effector immune responses. Previous studies have proposed that adequate induction of CD8+ effector T-cell responses by DCs (the “license to kill” theory) can be instructed by CD4 helper T cells that provide a maturation stimulus to DCs via CD40L acting on CD40, a system that can be replaced by systemic injection of CD40 agonistic antibodies.29, 30, 31 Therefore, recipients of OT-I cells were immunized IP with antibody/OVA conjugates with or without agonistic CD40 antibody. After 3 days, MLN cells from recipient mice were stimulated with phorbyl myristate acetate and ionomycin for 4 h, and interferon-γ (IFN-γ) production by OT-I cells was assessed by flow cytometry. IFN-γ-producing OT-I cells were abundant in MLNs of mice immunized with M290.OVA plus αCD40, but not in mice given M290.OVA alone or GLIII/10.OVA plus αCD40 (Figure 4a). To determine whether this immunization regime led to the generation of cytotoxic CD8+ T cells,32 C57BL/6 or CD103−/− mice were injected IP with antibody/OVA complexes with or without αCD40, and killing of adoptively transferred OVA-pulsed target cells was assessed in the spleen 12 days later (Figure 4b,c). Immunization with M290.OVA plus αCD40, but not M290.OVA alone or GLIII/10.OVA plus αCD40, induced the generation of cytotoxic T lymphocytes (CTLs). Again, this response was dependent on targeting to CD103+ cells, as it was not observed in CD103−/− mice (Figure 4b,c).

CD103+ dendritic cells (DCs) generate antigen-specific cytotoxic T lymphocytes (CTLs) and antibody responses in the presence of adjuvant. (a) M290.OVA plus αCD40 alone induces interferon-γ (IFN-γ) production in responding OT-I cells. Recipients of OT-I cells were injected intraperitoneally (IP) with M290.OVA (5 μg) or GLIII/10.OVA (15 μg) with/without αCD40. At 3 days after immunization, mesenteric lymph node (MLN) cells were isolated and restimulated with phorbyl myristate acetate (PMA) and ionomycin and intracellular expression of IFN-γ in OT-I cells was determined by flow cytometry. Mean (s.e.m.) of 5–6 mice per group from two separate experiments. (b, c) M290.OVA plus αCD40 induces antigen-specific CTL. M290.OVA (10 μg) or GLIII/10.OVA (20 μg) with/without αCD40 were injected IP into wild-type (WT) or CD103−/− mice. After 12 days, animals received peptide-pulsed (CFSElo) and unpulsed (CFSEhi) target cells and CTL activity was assessed in the spleen the following day by flow cytometry. (b) Representative flow cytometry plots and (c) mean (s.e.m.) of 5–9 mice per group from three experiments. ***P<0.001. (d) M290.OVA and adjuvant induces ovalbumin (OVA)-specific antibody responses. Mice were immunized IP with M290.OVA (filled symbols) alone (2.5 μg, square) or together with lipopolysaccharide (LPS; 50 μg, triangle) or polyinosinic:polycytidylic acid (polyI:C; 50 μg)/αCD40 (50 μg) (diamond). Control mice received only LPS (clear triangle) or polyI:C/αCD40 (clear diamond). After 4 weeks, mice received an IP injection with OVA (5 μg) and 6 days later serum levels of OVA-specific IgG1 and IgG2a was determined by enzyme-linked immunosorbent assay (ELISA). Data are the mean (s.e.m.) from one representative experiment of three (M290.OVA and LPS) or one (M290.OVA and polyI:C/αCD40) performed with three mice per group per experiment. Immunization with M290.OVA and LPS and M290.OVA and polyI:C/αCD40 induced significant IgG1 and IgG2a responses compared with M290.OVA alone (P<0.0001) as determined by two-way analysis of variance (ANOVA) test.
Coadministration of LPS or αCD40/polyinosinic:polycytidylic acid (poly I:C) together with antibody/OVA targeting complexes has previously been shown to induce antigen-specific antibody responses in vivo.33 To assess whether coadministration of these adjuvants with M290.OVA could induce antigen-specific antibody responses, C57BL/6 mice were injected with M290.OVA alone or together with αCD40/poly I:C or LPS. After 4 weeks, mice were boosted by IP injection of OVA and 6 days later serum levels of OVA-specific antibodies were determined by enzyme-linked immunosorbent assay. Coinjection of LPS with M290.OVA led to the generation of OVA-specific IgG1 and IgG2a, whereas administration of M290.OVA or adjuvant alone gave no response (Figure 4d). These results were confirmed using αCD40/poly I:C as combined adjuvants (Figure 4d). Together, these results suggest that CD103+ DCs in the presence of appropriate stimuli can prime immunity.
The absence of CTL or IFN-γ expression in CD8+ T cells in mice receiving M290.OVA alone, despite induction of CD8+ T-cell division, indicated that injection of M290.OVA to target CD103+ DCs in the steady state may induce tolerance in responding CD8+ T cells. To address this, recipients of OT-I cells were injected IP with M290.OVA with or without αCD40 or with phosphate-buffered saline (PBS) alone. After 12 days, mice received OVA in Complete Freund's Adjuvant (CFA) IP and 3 days later mice received an equal ratio of OVA peptide-pulsed (CFSElo) and unpulsed (CFSEhi) splenocyte targets. OVA peptide-pulsed splenocytes were selectively lysed in the MLNs and spleen of mice receiving PBS, or antibody/OVA conjugates together with αCD40 (Figure 5a,b), reflecting the induction of antigen-specific CD8+ CTL effectors by the OVA/CFA immunization. In sharp contrast, peptide-pulsed splenocyte targets in mice receiving only M290.OVA remained largely viable, despite immunization with CFA/OVA (Figure 5a,b). Thus, CD103+ DCs induce CTL tolerance in the absence of surrogate T-cell contact.

CD103+ dendritic cells (DCs) induces tolerance of CD8+ T-cell responses in the steady state. (a, b) OT-I cells were injected into recipient mice, and 1 day later recipients received M290.OVA (10 μg) with/without αCD40 (25 μg) or GLIII/10.OVA (20 μg) intraperitoneally (IP). At 12 days after immunization, mice were injected IP with ovalbumin (OVA; 50 μg) in Complete Freund's Adjuvant (CFA). After 3 days, carboxyfluorescein succinimidyl ester (CFSE)-labeled peptide-pulsed (CFSElo) and unpulsed (CFSEhi) splenocytes were injected intravenously (IV) and the killing of peptide-pulsed target cells assessed by flow cytometry. (a) Representative flow cytometry plots of CFSE-labeled splenocytes and (b) specific lysis of target cells in the mesenteric lymph nodes (MLNs). Results are the mean (s.e.m.) of 5–10 mice per group in three independent experiments. ***P<0.001.
Mucosal intratracheal delivery of M290.OVA induces T-cell proliferation in draining mediastinal LNs and tolerance to inhaled antigen
The above data suggested that antigen presentation by CD103+ DCs in the steady state leads to tolerance or immunity in the CD8+ T-cell compartment. However, all experiments were performed using systemic IP injection of M290.OVA, potentially also targeting DCs that are already resident in the LNs or spleen at the time of injection. To address the function of peripheral CD103+ DCs, we focused our attention to the lung, as antibodies can be injected into the trachea without the risk of being degraded and without mechanical breaching of physiological barriers. In the lung, CD103+ DCs are closely associated with pulmonary epithelial cells, extending their dendrites into the airway lumen and represent a major population of migratory DCs.6, 8, 11 To assess whether mucosal application of M290.OVA could be utilized to target tissue-resident CD103+ DCs, recipients of OT-I and OT-II cells received antibody/OVA conjugates by intratracheal (IT) injection (Figure 6a). Administration of as little as 28 ng of M290.OVA was sufficient to induce both OT-I and OT-II proliferation in draining mediastinal LNs (Figure 6a and Supplementary Figure S6a online), demonstrating remarkable targeting efficiency of this reagent across the lung mucosa.

Intratracheal (IT) administration of M290.OVA induces T-cell proliferation in mediastinal lymph nodes (LNs) and CD4+ T-cell mediated tolerance. (a) IT administration of M290.OVA induces OT-I and OT-II proliferation in mediastinal LNs. Carboxyfluorescein succinimidyl ester (CFSE)-labeled OT-I and OT-II cells were injected into recipient mice and proliferation assessed in mediastinal LNs 3 days after IT administration of M290.OVA (28 ng) or GLIII/10.OVA (56 ng) or ovalbumin (OVA) alone (56 ng). Results are representative histogram and dot plots (inserts) gating on donor OT-I and OT-II cells from one representative experiment of three performed with three mice per experiment. Numbers represent percentage of dividing cells (cells in red-lined box) within each population. (b–d) Mucosal application of M290.OVA induces tolerance. At 7 days after IT administration of M290.OVA (28 ng), GLIII/10.OVA (56 ng) or OVA (100 μg) mice were immunized with OVA and alum intraperitoneally (IP) followed by OVA-aerosol challenges. IL, interleukin; PBS, phosphate-buffered saline. (b) Immune cell populations in bronchoalveolar lavage fluid (BALF). (c) Cytokine analysis in mediastinal LN preparations 3 days after in vitro restimulation with OVA. (d) Immunoglobulin E (IgE) levels in serum. Results are from one representative experiment of three performed. *P<0.05; **P<0.01.
We next utilized this system to study the functional outcome of mucosal application of M290.OVA, focusing specifically on mucosal tolerance. To address induction of inhalation tolerance, mice received antibody/OVA complexes IT, and 7 days later were immunized IP with OVA and the T helper type 2 (Th2) adjuvant alum. After 10 days, mice were challenged for 3 consecutive days with 1% OVA aerosol to elicit allergic type airway inflammation. At 24 h after the last challenge, bronchoalveolar lavage (BAL) fluid, mediastinal LN, and serum were collected to address the induction of asthma features. In mice injected with PBS before immunization with OVA/alum and aerosol exposure, there was induction of airway eosinophilia (Figure 6b), accompanied by Th2 cytokine production in the mediastinal LN lymphocytes (Figure 6c), a rise in serum OVA-specific immunoglobulin E (IgE) levels (Figure 6d) as well as inflammatory peribronchiolar and perivascular infiltrates (Supplementary Figure S6b online). These features of allergic asthma were severely reduced when mice received a single injection of 100 μg of OVA intratracheally before the immunization protocol, demonstrating the effect of inhalation tolerance induction. The IT administration of M290.OVA but not GLIII/10.OVA also significantly attenuated airway eosinophilia, mediastinal LN Th2 cytokine production, and anti-OVA IgE antibody responses to OVA/Alum, to the degree observed in mice receiving 100 μg of soluble OVA. Therefore, targeting OVA specifically to mucosal CD103+ lung DCs is highly efficient in inducing inhalation tolerance.
M290 targets distinct and partially overlapping subsets of DCs compared with DEC205 and 33D1 antibodies
Targeted delivery of antigen to DCs in vivo has therapeutic potential in vaccine design and for antitumor and disease-modulation therapies.34 Two targets on DCs that have received particular attention are the C-type lectin receptors DEC205 and DCIR2 (recognized by the monoclonal antibody 33D1) that are expressed on splenic CD8+ and CD8− DCs, respectively.2, 31, 35 To determine whether the DC subsets targeted by CD103 are similar or distinct from those targeted by DEC205 and 33D1, expression of DEC205 and 33D1 on CD103+ and CD103− MLNs, mediastinal LNs, and splenic DCs was assessed by flow cytometry (Figure 7). DEC205 was expressed on the majority of CD103+ DCs in MLNs (80%), mediastinal LNs (85%), and spleen (83%), as well as on a large proportion of CD103− DCs (Figure 7a–c). In contrast, 33D1 was expressed on ∼40% of CD103+ DCs in MLNs and on only a minor fraction of mediastinal LNs (5%) and splenic CD103+ DCs (34%), but on a sizeable fraction of CD103− DCs in all three tissues (Figure 7a–c). Thus, M290 targets a distinct array of DCs compared with DEC205 and 33D1.

Expression of DEC205 and 33D1 on CD103+ and CD103− dendritic cells (DCs). Expression of DEC205, 33D1, and CD103 expression on (a) mesenteric lymph node (MLN), (b) mediastinal LN (Med. LN), and (c) splenic lineage (CD3, CD19, NK1.1)−, CD11c+ MHCII+ DCs. Numbers represent the mean (s.d.) percentage of positive cells compared with isotype control (shaded). Results are from one representative staining of three performed using three (pooled for med. LN) mice per experiment.
Discussion
CD103+ DCs represent a major subset of migratory DCs in peripheral tissues including the lung, intestine, and dermis; however, their in vivo functional activities remain to be fully elucidated. Here, by conjugating OVA to the CD103 antibody, M290, we were able to directly address the priming potential of CD103+ DCs in vivo.
Our results show that CD103+ DCs in MLN, spleen, and mediastinal LNs are capable of stimulating both CD4+ and CD8+ T-cell responses in vivo. Administration of M290.OVA alone induced antigen-specific tolerance, suggesting that CD103+ DCs promote tolerogenic responses in the steady state. However, coadministration of adjuvant switched this tolerogenic activity to one of immunity, leading to the generation of antigen-specific CTLs and antibody responses. These results demonstrate that CD103+ DCs, when appropriately activated, can be induced to promote effector responses in vivo. Although from these in vivo studies, we could not determine whether CD103+ DCs residing in different locations differed in their immune-promoting activities, these results are consistent with recent, primarily in vitro, observations that even intestinal CD103+ DCs when appropriately stimulated can switch from a tolerance to immune-promoting phenotype.36, 37, 38
Consistent with previous in vitro findings,4, 16, 19 CD103+ MLN DCs efficiently induced gut-homing receptor expression on OT-I cells in vivo; however, surprisingly little CCR9 was induced on OT-II cells. Although the reason for inefficient CCR9 induction on CD4+ T cells is currently unclear, we consistently observe less CCR9 induction on OT-II cells compared with OT-I cells following oral or IP administration of OVA (M. Svensson-Frej, M. Semmrich, unpublished observation; B. Johansson-Lindbom, personal communication), indicating that such differences are not due to the M290 targeting strategy. Gut-homing receptor induction on T cells in the MLN in vivo and by CD103+ DCs in vitro is dependent on the Vitamin A (retinol) metabolite retinoic acid.19, 20, 39 Thus, one possibility is that CD4+ T-cell priming in vivo occurs in a less retinoic acid-rich environment. For example, different subsets of CD103+ MLN DCs may preferentially prime CD4+ and CD8+ T cells in vivo, and the CD103+ DC subset that primes CD4+ T cells may be less efficient at generating retinoic acid. Alternatively, as subsets of MLN stromal cells generate retinoic acid,40 the MLN stromal environment in which CD4+ and CD8+ T-cell priming takes place may also differ. A second possibility is that CD4+ T cells receive additional competing differentiation signals in vivo. In this regard, CXCR5+ follicular helper OT-II cells develop in parallel to gut-homing OT-II cells in the MLNs following IP administration of OVA and adjuvant.41 Finally, antigen dose is an important parameter in regulating tissue homing receptor expression at least on OT-I cells,19 and thus the amount of antigen presented on major histocompatibility complex class I (MHC-I) and MHC-II may influence the efficiency of gut-homing receptor induction.
In addition to their ability to induce gut-homing receptors, CD103+ MLN DCs have received considerable attention because of their enhanced capacity to promote FoxP3+ T-cell differentiation in vitro, a property that appears, at least in part, to be mediated through their ability to generate transforming growth factor-β and retinoic acid.16, 17, 42, 43 We were thus initially surprised that in vivo targeting of antigen to CD103+ DCs did not induce higher OT-II cell conversion to FoxP3+ Tregs in the MLNs in vivo. Nevertheless, we and others have previously demonstrated that CD103+ DCs play a central role in picking up and presenting oral antigen to CD4+ T cells in the MLNs,5, 16 and under similar conditions, only 1–8% of CD4+ T cells convert to FoxP3+ Tregs in the MLNs.16, 17, 28 In addition, in the absence of exogenous transforming growth factor-β, FoxP3+ Treg conversion by CD103+ DCs in vitro also lies within this range (2.5–8%).16, 17 Together, these findings suggest that CD103+ DC-mediated FoxP3+ Treg conversion in MLNs in vivo is rather inefficient and are in line with a recent study suggesting that the expansion of newly differentiated FoxP3+ Tregs occurs primarily in the SI-LP.28
The possibility of selectively targeting antigen to mucosal DCs by mucosal application of antibody/antigen complexes has not to our knowledge been previously assessed. Here, we demonstrate that IT administration of M290.OVA but not GLIII.10.OVA was extremely efficient at inducing antigen-specific CD4+ and CD8+ T-cell proliferation in draining mediastinal LNs. The ability of lung-derived CD103+ DCs to prime CD4+ T cells contrasts with a report from del Rio et al.15 indicating that these cells, while efficiently cross-presenting soluble antigen, are poor at priming CD4+ T cells, although they are consistent with the observation that mediastinal LN CD103+ DCs from mice infected with OVA-expressing influenza virus prime OVA-specific CD4+ T cells ex vivo.8 Notably, despite inducing antigen-specific CD4 and CD8 T-cell proliferation, the outcome of IT M290.OVA administration was inhalation tolerance as measured by reduced levels of antigen-specific IgE, Th2 responses, and eosinophil accumulation in the BAL fluid. Although more studies are needed to determine the mechanism(s) by which this subset of DCs induce tolerance, for diseases like asthma, one can envisage tolerizing protocols exploiting the steady-state immunoregulatory potential of mucosal CD103+ DCs to induce more long-lasting prevention of disease features.
Specific targeting of antigen to DCs in vivo with antibodies or ligands has proven a feasible means of enhancing adaptive immune responses and has great potential in the field of vaccination therapy.34 Although targeting strategies have tended to focus on the C-type lectin receptor family, including DEC205, DC-SIGN, Mannose receptor, CLEC9A, and DCIR2,34, 44, 45 antibody-mediated targeting to CD11c was also recently shown to be highly effective in the generation of CD4+ and CD8+ T-cell responses and antitumor immunity.46, 47 Our demonstration that CD103 can function as an uptake receptor on DCs and induce antigen-specific CD4+ and CD8+ T-cell priming in vivo further expands the array of potential receptors that may be utilized for the targeted delivery of antigen and/or immunomodulatory substances to DCs in vivo. Our results suggest, at least in mouse, that CD103 is expressed on a partially overlapping, and partially distinct set of DCs compared with DEC205+ and 33D1+ DC subsets. Although additional studies are required to directly compare these targeting strategies in different immunological models, we hypothesize, based on the accumulated evidence that distinct DC subsets display different functional characteristics, that targeting CD103+ DCs (either via CD103 or other receptors on these cells) may prove advantageous in certain settings. In this regard, CD103+ DCs with similar function and phenotype have been identified in human MLNs and are also present in the intestinal lamina propria and skin-draining LNs.5, 18, 48, 49 Furthermore, in contrast to the mouse,25 the majority of naive human CD8+ T cells do not express CD103.50
Methods
Mice. C57BL/6, OT-I.CD45.1, OT-II, OT-II.CD45.1, C57BL/6.CD45.1, and CD103−/− mice were bred and maintained at the Biomedical Center Animal Facility (Lund University, Lund, Sweden) and used between 6 and 14 weeks of age. CD11c.DTR mice were bred and maintained at the Experimental Biomedicine Animal Facility, University of Gothenburg, Sweden. All animal experiments were approved by the Lund/Malmö animal ethics committee.
Antibodies and reagents. The following antibodies and reagents were used during the course of this study: PE-CD103 (M290), PE-α4β7 (DATK32), PE-CD11c (N418), bio-CD45.1 (A20), APC-CD8α (53–6.7), PE-Vβ (MR9-4), bio-Vα2 (B20.1), FITC or bio-CD8β (53-5.8), PE or bio-rat IgG2a (RG7/1.30), bio-rat IgG2b (G15-337), and anti-rat IgG-APC (all from BD Bioscience, Franklin Lakes, NJ); pacific blue-MHC class II (M5/114.15.2), Alexa700-CD45.1 (A20), Alexa700-CD45.2 (104), CD40 (1C10), and PE-Cy5-NK1.1 (PK136) (all from BioLegend, San Diego, CA); bio-CD103 (2E7), bio- or pacific blue-CD8α (53-6.7), PE-Cy7-CD11c (N418), PE or PE-Cy7-B220 (RA3-6B2), PE-Cy7-CD4 (GK1.5), pacific blue-CD4 (RM4-5), IFN-γ–PE-Cy7 (XMG1.2), APC-FoxP3 (FJK16s), Alexa Fluor-647-CD107a (eBio1D4B), PE-Cy7-CD25 (P61.5), PE-Cy5-CD3 (145-2C11), PE-Cy5-CD19 (eBio1D3), bio-33D1 (33D1), streptavidin-conjugated APC, and the Foxp3 staining kit (all from eBioscience, San Diego, CA). Anti-EEA-1 (rabbit polyconal) was from Cell Signaling Technology (Danvers, MA). Bio-DEC-205 (NLDC-145) was from Abcam (Cambridge, UK). Cy3-conjugated donkey anti-rabbit IgG-F(ab’)2 was from Jackson Immunoresearch Laboratories (West Grove, PA). Anti-CCR9 antibody,51 anti-CD103 (M290), and rat IgG2a isotype-control (GLIII/10) were purified from hybridomas. Alexa Fluor-647 protein-labeling kit was from Invitrogen (Carlsbad, CA). OVA grade VII, LPS (Escherichia coli, serotype 055:B55), DNase I, collagenase type IV and VIII, CFA, and CFSE were from Sigma-Aldrich (St Louis, MO). FTY720 was obtained from Cayman Chemical (Ann Arbor, MI). Synthetic pOVAs (OT-I, SIINFEKL and OT-II, ISQAVHAAHAEINEAGR) were from Innovagen (Lund, Sweden).
Cell isolation. Intraepithelial lymphocytes and lamina propria cells were isolated as previously described.25, 52 For isolation of LN T cells and DCs, LNs were cut and filtered directly through a 70 μm cell strainer or incubated with collagenase IV (500 μg ml–1) and DNAse I (50 U ml–1) for 30 min at 37 °C before filtering, respectively. CD4+ and CD8β+ T cells were enriched to >85% purity by magnetic-activated cell sorting (MACS; Miltenyi Biotech, Bergisch Gladbach, Germany) using CD4 MACS beads or bio-anti-CD8β (53-5.8) followed by streptavidin-conjugated MACS beads, respectively. In Figure 1a and Supplementary Figure S2b and c, DCs were isolated from mice injected with Flt3L (FMS-like tyrosine kinase-3 ligand)-secreting B16 melanoma cells.
Flow cytometry and cell sorting. Flow cytometry was performed according to standard procedures. Dead cells were identified using propidium iodide staining or Live Dead Fixable Violet/Red Dead cell staining kit (Invitrogen) and removed from the analysis. Data acquisition was performed on a FACSCalibur, FACS LSRII, or FACSAria (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR). For purification of CD103+ and CD103− DCs, MACS-enriched CD11c+ DCs were stained with anti-CD11c, anti-CD103, and anti-MHC class II and sorted on a FACSAria.4 DC fractions were routinely 95–99% pure, respectively.
Immunohistochemistry. Tissue sections (7 μm) were fixed in 4% paraformaldehyde. After blocking with rat and goat serum (10% of each in PBS, 0.05% Tween), sections were incubated with anti-CD11c (N418), followed by anti-B220-FITC (RA3-6B2) and Cy3 goat anti-hamster IgG (polyclonal, Jackson ImmunoResearch).
Antibody uptake assay. MLN DCs were isolated using CD11c microbeads (Miltenyi Biotec) and were 90% CD11c+MHC class II+. DCs were labeled with biotinylated M290 or isotype control for 30 min on ice followed by labeling with Alexa-488 conjugated streptavidin (Invitrogen) for 15 min on ice. The cells were washed in medium and incubated at 37 °C for indicated time periods. For intracellular staining of lysosomes and endosomes, DCs were fixed and permeabilized with Leucoperm (AbD Serotec, Kidlington, UK) and stained with Alexa-647 conjugated anti-CD107a (LAMP-1) and rabbit anti-EEA1mAb followed by anti-rabbit IgG-Cy3. Cells were centrifuged on coverslips and mounted in ProLong Gold anti-fade reagent (Invitrogen) and imaged with an epifluorescent microscope (Axiovert 200M; Carl Zeiss AB, Stockholm, Sweden) equipped with a × 63 1.4 Plan-Apochromat oil objective and Hamamatsu Orca-ER camera. Images were acquired at 0.3 μm steps spanning the entire cell, deconvolved by fast restoration, and three-dimensional reconstructions prepared using Volocity software 5.2.1 (Perkin Elmer, Waltham, MA).
Generation of antibody/OVA conjugates. M290 and GLIII/10 were conjugated to maleimide-activated OVA (Pierce Biotechnology, Rockford, IL) according to the manufacture's protocol. Briefly, antibodies were reduced with 2-mercaptoethylamine HCl (MEA) and separated from MEA over a desalting column. Maleimide-activated OVA and reduced antibodies were incubated in a 1:1 molar ratio for 2 h at room temperature. Samples were run over a protein G column (Pierce Biotechnology) to remove unbound OVA. Western blot was performed to confirm conjugation and removal of unbound OVA and enzyme-linked immunosorbent assay was performed according to standard techniques to determine the amount of OVA conjugated to each antibody. GLIII/10.OVA conjugates consistently contained less OVA than M290.OVA conjugates (approximately twofold less) and the concentrations used for experiments adjusted accordingly.
In vitro proliferation assay. DCs were pulsed with antibody/OVA conjugate for 3 h, washed extensively, and incubated in a 1:2 ratio with OT-I cells (2 × 105) in 96-well flat-bottom plates (Thermo Fisher Scientific, Waltham, MA) in 200 μl R10-medium (RPMI-1640, 10% fetal calf serum, 10 mM Hepes, 1 mM sodium pyruvate, 50 μM mercaptoethanol, 100 U ml–1 penicillin, 100 μg ml–1 streptomycin, and 50 μg ml–1 gentamicin; all from Invitrogen). Cell proliferation was determined by adding 1 μCi [3H]-thymidine (Amersham Biosciences, Uppsala, Sweden) to the cultures at 24 h and assessing thymidine incorporation 16 h later.
In vivo T-cell priming. MACS-purified OT-I.CD45.1 and OT-II.CD45.1 cells were labeled with CFSE according to standard protocols and injected (3–5 × 106) intravenously into C57BL/6 or CD103−/− mice. To determine the generation of FoxP3+ CD4+ T cells, naive CD4+CD44loCD25−FR4−CD69− were sorted from OT-II mice before their transfer into recipient animals. For IT immunization, MACS-purified OT-I.CD45.1 or OT-II.CD45.1 cells were labeled with CFSE according to standard protocols and injected (3–5 × 106) intravenously into C57BL/6. After 24 h, recipients were immunized IT with antibody/OVA conjugates at the indicated dose. In some experiments, mice received FTY720 (1 mg kg–1 body weight; Cayman, Talinn, Estonia) IP 24 h later. For DC depletion, CD11c-DTR mice were injected IP with 4 ng g–1 body weight DTx at the time of OT-I and OT-II T cell transfer. Recipients were immunized IP with antibody/OVA conjugates at the indicated dose.
In vivo cytotoxicity assay. Splenocytes were incubated with/without SIINFEKL peptide (0.2 μM) for 2 h at 37 °C, washed, and labeled with low (0.2 μM) and high (2 μM) doses of CFSE, respectively. After washing, labeled cells were mixed in a 1:1 ratio and intravenously injected into recipient animals (6 × 106 cells per mouse). After 17 h, spleens and MLN cell suspensions from the recipients were stained and analyzed by flow cytometry. The percentage of killing was quantified as (1−(ratio unprimed/ratio primed)) × 100, with ratio determined as %CFSE nonpulsed/ %CFSE pulsed cells. For intracellular cytokine analysis, MLN cells from recipients of OT-I cells were isolated 3 days after immunization, and restimulated with phorbyl myristate acetate (100 ng ml–1; Sigma-Aldrich) and ionomycin (1 μg ml–1; Sigma-Aldrich). Brefeldin A (20 μg ml–1; Sigma-Aldrich) was added after 1 h, cells were harvested 4 h later, and stained for intracellular IFN-γ according to standard techniques.
In vivo tolerance model. C57BL/6 mice were immunized IT with antibody/OVA conjugates, OVAwortington (100 μg), or PBS as control. After 7 days, mice were injected IP with 500 μl of Imject alum suspension (1 mg) containing 10 μg of OVA (OVA-alum). After 10 days, mice were challenged with 1% OVA GIII (Sigma, St Louis, MO) aerosols for 3 consecutive days. At 24 h after the last challenge, serum, BAL, and mediastinal LNs were obtained. Enzyme-linked immunosorbent assay was performed to measure antibody response in serum and cytokine production from mediastinal LNs (eBioscience) after restimulation for 3 days with OVA (20 μg ml–1). To determine the cellular composition of BAL fluids, cells were stained with MHCII-FITC (2G9; for DC and macrophage staining), CD11c-APC (HL3; for DC staining), CD19-CyChr (RA3-6B2) and CD3-CyChr (145-2C11; BD), and Siglec F-PE (R&D Systems, Abingdon, UK; for eosinophil staining). The cellular composition of BAL fluid was determined using flow cytometry. For histology sections, lungs were inflated using OCT (optimal cutting temperature compound)/PBS and frozen. Subsequently, cryosections were made and stained with periodic acid-Schiff reagent (Sigma) and photographed with an Olympus BX53 microscope (Olympus, Center Valley, PA).
Statistical analysis. Unless otherwise mentioned, statistical analyses were performed using unpaired or paired two-tailed Student's test. *P<0.05; **P< 0.01; and ***P<0.001.
References
den Haan, J.M. & Bevan, M.J. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(−) dendritic cells in vivo. J. Exp. Med. 196, 817–827 (2002).
Dudziak, D. et al. Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111 (2007).
Merad, M. & Manz, M.G. Dendritic cell homeostasis. Blood 113, 3418–3427 (2009).
Johansson-Lindbom, B. et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 202, 1063–1073 (2005).
Jaensson, E. et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 205, 2139–2149 (2008).
Sung, S.S. et al. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176, 2161–2172 (2006).
Bursch, L.S. et al. Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med. 204, 3147–3156 (2007).
GeurtsvanKessel, C.H. et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 205, 1621–1634 (2008).
Poulin, L.F. et al. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J. Exp. Med. 204, 3119–3131 (2007).
Ginhoux, F. et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J. Exp. Med. 204, 3133–3146 (2007).
Hintzen, G. et al. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J. Immunol. 177, 7346–7354 (2006).
Henri, S. et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 207, 189–206 (2010).
Bedoui, S. et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat. Immunol. 10, 488–495 (2009).
Desch, A.N. et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 208, 1789–1797 (2011).
del Rio, M.L., Rodriguez-Barbosa, J.I., Kremmer, E. & Forster, R. CD103- and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178, 6861–6866 (2007).
Coombes, J.L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Sun, C.M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Iliev, I.D. et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut 58, 1481–1489 (2009).
Svensson, M. et al. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol. 1, 38–48 (2008).
Iwata, M. et al. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538 (2004).
Zhang, L. et al. An anti-CD103 immunotoxin promotes long-term survival of pancreatic islet allografts. Am. J. Transplant. 9, 2012–2023 (2009).
Mahnke, K. et al. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell. Biol. 151, 673–684 (2000).
Kaden, S.A. et al. Enhanced dendritic cell-induced immune responses mediated by the novel C-type lectin receptor mDCAR1. J. Immunol. 183, 5069–5078 (2009).
Kilshaw, P.J. & Murant, S.J. A new surface antigen on intraepithelial lymphocytes in the intestine. Eur. J. Immunol. 20, 2201–2207 (1990).
Svensson, M. et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Invest. 110, 1113–1121 (2002).
Annacker, O. et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061 (2005).
Voedisch, S. et al. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of Salmonella enterica serovar Typhimurium and limit systemic disease in mice. Infect. Immun. 77, 3170–3180 (2009).
Hadis, U. et al. Intestinal tolerance requires gut homing and expansion of FoxP3(+) regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011).
Schoenberger, S.P., Toes, R.E., van der Voort, E.I., Offringa, R. & Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393, 480–483 (1998).
Bennett, S.R. et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480 (1998).
Bonifaz, L. et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638 (2002).
Yang, Y., Huang, C.T., Huang, X. & Pardoll, D.M. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 5, 508–515 (2004).
Boscardin, S.B. et al. Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J. Exp. Med. 203, 599–606 (2006).
Steinman, R.M. Dendritic cells in vivo: a key target for a new vaccine science. Immunity 29, 319–324 (2008).
Hawiger, D. et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–779 (2001).
Siddiqui, K.R., Laffont, S. & Powrie, F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 32, 557–567 (2010).
Fujimoto, K. et al. A new subset of CD103+CD8alpha+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces Th1 response and CTL activity. J. Immunol. 186, 6287–6295 (2011).
Uematsu, S. et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9, 769–776 (2008).
Jaensson-Gyllenback, E. et al. Bile retinoids imprint intestinal CD103(+) dendritic cells with the ability to generate gut-tropic T cells. Mucosal Immunol. 4, 438–447 (2011).
Hammerschmidt, S.I. et al. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 205, 2483–2490 (2008).
Cucak, H., Yrlid, U., Reizis, B., Kalinke, U. & Johansson-Lindbom, B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity 31, 491–501 (2009).
Siddiqui, K.R. & Powrie, F. CD103+ GALT DCs promote Foxp3+ regulatory T cells. Mucosal Immunol. 1 (Suppl 1), S34–38 (2008).
Belkaid, Y. & Oldenhove, G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 29, 362–371 (2008).
Tacken, P.J. & Figdor, C.G. Targeted antigen delivery and activation of dendritic cells in vivo: steps towards cost effective vaccines. Semin. Immunol. 23, 12–20 (2011).
Shortman, K., Lahoud, M.H. & Caminschi, I. Improving vaccines by targeting antigens to dendritic cells. Exp. Mol. Med. 41, 61–66 (2009).
Castro, F.V. et al. CD11c provides an effective immunotarget for the generation of both CD4 and CD8 T cell responses. Eur. J. Immunol. 38, 2263–2273 (2008).
Wei, H. et al. Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin. Cancer Res. 15, 4612–4621 (2009).
Beitnes, A.C. et al. Density of CD163(+) CD11c(+) dendritic cells increases and CD103(+) dendritic cells decreases in the coeliac lesion. Scand. J. Immunol. 74, 186–194 (2011).
van de Ven, R. et al. Characterization of four conventional dendritic cell subsets in human skin-draining lymph nodes in relation to T-cell activation. Blood. 118, 2502–2510 (2011).
McFarland, R.D., Douek, D.C., Koup, R.A. & Picker, L.J. Identification of a human recent thymic emigrant phenotype. Proc. Natl. Acad. Sci. USA 97, 4215–4220 (2000).
Pabst, O. et al. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J. Exp. Med. 199, 411–416 (2004).
Schulz, O. et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 206, 3101–3114 (2009).
Acknowledgements
We thank Dr R. Hodes (NCI, Rockville) for providing the GLIII/10 hybridoma, Drs G. Butcher and P. Kilshaw (Babraham Institute, Cambridge) for providing the M290 hybridoma, and Ann-Charlotte Selberg for valuable technical assistance. This work was supported by grants from the Swedish Medical Research Council, the Wellcome Trust, the Crafoordska, Torsten and Ragnar Söderbergs, Kocks, Österlund and Richard and Ruth Julins foundations, the Royal Physiographic Society, and the Swedish foundation for Strategic Research FFL-2 program. M.S. was supported by Deutsche Forschungsgemeinschaft Grant SE 1874/1-1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Semmrich, M., Plantinga, M., Svensson-Frej, M. et al. Directed antigen targeting in vivo identifies a role for CD103+ dendritic cells in both tolerogenic and immunogenic T-cell responses. Mucosal Immunol 5, 150–160 (2012). https://doi.org/10.1038/mi.2011.61
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2011.61
This article is cited by
-
ADP-ribosylating adjuvant reveals plasticity in cDC1 cells that drive mucosal Th17 cell development and protection against influenza virus infection
Mucosal Immunology (2022)
-
ADAMTS7 Attenuates House Dust Mite-Induced Airway Inflammation and Th2 Immune Responses
Lung (2022)
-
The role of β2 integrin in dendritic cell migration during infection
BMC Immunology (2021)
-
Lung IFNAR1hi TNFR2+ cDC2 promotes lung regulatory T cells induction and maintains lung mucosal tolerance at steady state
Mucosal Immunology (2020)
-
Neonatal mice possess two phenotypically and functionally distinct lung-migratory CD103+ dendritic cell populations following respiratory infection
Mucosal Immunology (2018)

{kind=link}
{kind=link}