
Abstract
Regulatory T cells (Treg) control an array of immune responses both in the context of various polarized settings as well as in distinct microenvironments. This implies that maintenance of peripheral homeostasis relies on the capacity of Treg to appropriately adapt to these defined settings while sustaining a regulatory program in the face of inflammation. Adaptation of Treg is particularly critical in tissues constantly exposed to microbes, such as the gut or the skin, or in the context of exposure to pathogenic microbes. Recent evidence supports the idea that the capacity of Treg to control defined polarized settings can be associated with the acquisition of specific transcription factors previously associated with effector T-cell lineages. In this review we will discuss how such adaptation of Treg can have a major role in the control of host–microbe interaction.
Similar content being viewed by others


A Role for T-bet in the Control of Regulatory T cells (Treg) Function at Th1 Sites
One required feature of tissue regulation relies on the proper accumulation of regulatory cells in the inflamed tissue. Until recently it was unclear how Treg responded to environmental cues and targeted defined sites. A recent report by Koch et al.1 supports the idea that T-bet expression by Treg may be instrumental in the capacity of Treg to accumulate at Th1 polarized sites. Using various experimental settings and in particular Mycobacterium tuberculosis infection, this group demonstrated that acquisition of T-bet via its capacity to induce CXCR3 favors the homing of Treg to Th1 sites of inflammation. In competitive bone marrow chimeras Tbx1−/− Treg cells were outcompeted by wild-type Treg during Th1 inflammation, suggesting an additional role for T-bet in their survival or proliferation. The induction of T-bet in Treg was found to be interferon gamma (IFN-γ) dependent, yet did not require expression of interleukin (IL)-12Rβ.1 Similarly, during Toxoplasma gondii infection, T-bet expression correlated with expression of CXCR3.2 When isolated from the primary site of T. gondii infection, small-intestine lamina propria dendritic cells (DCs) readily induced T-bet expression by Treg, owing, in part, to their capacity to induce IFN-γ by T cells. As LpDCs gained the capacity to produce IL-12 in this environment, T-bet expression was associated with acquisition of responsiveness to IL-12 via enhanced Stat4 (signal transducer and activator of transcription 4) phosphorylation.2 Other factors—such as IL-27 highly expressed in lamina propria DCs from infected mice—are also likely to contribute to this imprinting. At the population level, the expression of T-bet did not interfere with the capacity of Treg to suppress proliferation of effector T cells in vitro.1, 2 Thus, the appropriation of T-bet seems to provide a fitness advantage to Treg in the context of Th1-polarized infections. Such control can be associated with an enhanced homing property as well as acquisition of responsiveness to defined growth factors such as IL-12 present in Th1-polarized microenvironments
Adaptation of Treg to Sites Constitutively Exposed to Microbes
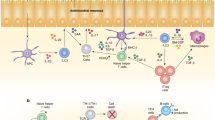
At steady state, the gut is home to a large number of lymphocytes that have the capacity to produce cytokines such as IL-17, IFN-γ and IL-4.3, 4 This constitutive production of cytokines is tightly controlled by the flora, as germ-free mice show extensive deficiencies in intestinal immune system development and basal cytokine production.3, 5 Based on the aforementioned findings, one can speculate that in order to control immune responses at mucosal sites Treg may express transcriptional programs analogous to tissue-resident effector T cells. Furthermore, such barrier surfaces may require more proficient Treg to maintain homeostasis. In support of this theory, previous studies have identified two other transcription factors, IRF46 and Stat3,7 associated with effector function and responsiveness, to be required for the capacity of Treg to control Th2 and Th17 inflammation, respectively. Zheng et al.6 first demonstrated that IRF4 expression by Treg was required to control Th2 pathology as mice with IRF4−/− Treg succumb to disease directed at multiple barrier sites, including the lungs, stomach, and pancreas, by 3–4 months of age. Similarly, Chaudhry et al.7 showed that selective deletion of Stat3 in Foxp3+ cells resulted in the development of an uncontrolled and lethal Th17 inflammation in the gut. Examination of the requirements for these transcription factors in polarized infectious settings will provide a more complete understanding of the capacity of Treg to adapt to defined microenvironments (Figure 1).

Features of tissue-resident regulatory T cells (Treg) during homeostasis and infection. The schematic depicts the stable expression of Foxp3 during thymic generation (and potentially de novo generation in the gut). As Treg enter the periphery, especially of tertiary tissues, the level of plasticity of Treg increases during homeostasis and especially infection.
Excessive Adaptation as a Potential Trigger of Immunopathology
Initial studies in T helper differentiation described a unidirectional pathway to effector lineage commitment and cytokine production. However, recent evidence has shown that lymphocytes maintain a certain degree of plasticity with respect to their capacity to produce cytokines.8, 9 Cells expressing both Foxp3 and IL-17 can be found in mucosal tissue or in vitro cultures.8, 10, 11 Genomewide mapping of H3K4me3 and H3K27me3 performed in ex vivo Treg revealed markers of both repression and induction at the tbx21 locus. On the other side, the IFN-γ locus did not show any sign of induction or repression,9 suggesting that it is poised for transcriptional activation. A previous report demonstrated that Treg expressing both Foxp3 and T-bet were induced by CD8α+ DCs and could protect against airway hyperactivity.12 A role for IFN-γ in mediating Treg function has been reported in a model of graft transplants13 and recent evidence demonstrates that, in vitro, Treg can acquire expression of this cytokine.9 Following oral infection with T. gondii under conditions associated with high immunopathology and eventual death of the infected host, Treg can produce IFN-γ, which is a cytokine responsible for both effector and pathogenic responses during this infection. When isolated from infected animals, Treg were able to exert effector functions as evidenced by their capacity to activate macrophages and induce parasite killing.2 Such an aberrant fate for Treg appears to be associated with, or to arise as a consequence of, pathology. Indeed, IFN-γ production by Treg was only detected in situations leading to death of the infected host. This would suggest that in the presence of high levels of inflammatory mediators, T-bet expression may reach a threshold that could lead to Treg destabilization. Notably, in T. gondii-infected mice, the level of T-bet in Treg was much higher than that observed in Treg during M. tuberculosis infection.1, 2 Both M. tuberculosis and T. gondii are strong Th1-inducing infections; however, under certain conditions, T. gondii triggers a cytokine storm, with very high levels of inflammatory cytokines such as IL-6, IL-27, and IL-12. This in conjunction with severely decreased levels of IL-2, which was also seen during this infection, may act on Treg to imprint an effector phenotype. Indeed, when isolated from the primary site of infection, lamina propria DCs from T. gondii-infected mice can only induce IFN-γ production by Treg in the presence of high levels of IL-12. This implies that acquisition of IFN-γ by these cells requires an amplification loop provided by enhanced IL-12 production, a response not normally seen in the gut environment.2
Another example of adaptation of Treg to microbes was observed in the context of exposure to fungal products. Treg exposed to DCs that had been incubated with curdlan, a β-glucan, co-express RAR-related orphan receptor gamma-t.14 A similar phenotype had been previously described on Treg residing in the intestinal mucosa.15 The physiological relevance of RAR-related orphan receptor gamma-t expression for Treg function remains to be addressed, but the observation that microbial products or exposure to sites exposed to microbes favors this phenotype suggests that, as for T-bet, RAR-related orphan receptor gamma-t may represent a positive adaptation of Treg cells in defined settings.
On the other hand, as observed with pathogenic levels of infection with T. gondii, high doses of curdlan led to the production of IL-17 by Foxp3+RAR-related orphan receptor gamma-t+ Treg, which was dependent on IL-23 production by DCs. Similarly, Treg resident in mucosal tissues of both mice and humans can produce IL-17.15, 16 The roles of IFN-γ or IL-17-producing Treg are difficult to assess. However, given their high degree of Treg self-reactivity, it is plausible that, if armed with effector cytokines, these cells can contribute to tissue damage or can lose their suppressive capacity. Indeed, a recent report highlighted that Foxp3 instability and acquisition of IFN-γ can favor the development of autoimmune diabetes.17
Control of Treg Conversion by a Defined Environment
In addition to the regulation provided by thymically derived Treg, recent findings support the idea that the gastrointestinal tract represents a privileged site for the induction of Treg from naïve CD4+ T cells. Previous work demonstrated that in vitro Treg conversion was abolished in the presence of Th1- or Th2-associated effector cytokines.18, 19, 20, 21 In addition, IL-6 required for polarization towards Th17 can down-modulate Foxp3 expression. Accordingly, conversion in the highly Th1 response to T. gondii is halted.2 Interestingly, converted Treg, although reduced in number during this infection, still adapt to the Th1 environment by expressing T-bet,2 suggesting that plasticity is not the sole prerogative of naturally occurring Treg cells. Previous reports examining both gut and lung inflammation support the idea that restricted or defective Treg conversion can enhance immunopathology.22, 23 The relative contribution of blockade of Treg conversion to the pathology induced by T. gondii remains difficult to evaluate but is likely to have a role in the overall decrease of Treg during this infection. These findings also raise the possibility that exposure to antigen at a time of acute infection may impair the acquisition of tolerance against innocuous antigens (e.g., flora or food antigens), which could, in turn, further contribute to the pathological process.
As highlighted by the studies discussed, plasticity of Treg during infection may have a positive role in their capacity to target defined sites, control polarized settings, and survive in a competitive manner with the cells they have to regulate. On the other hand, acquisition of additional transcription factors may lead to Treg destabilization with acquisition of effector cytokines and, in some cases, loss of Foxp3. Another important point to consider is that in most cases strict polarization of immune responses is a rare event in tissues. How Treg integrate these complexes and in some cases antagonistic signals to adapt appropriately remains to be addressed. A further examination of Treg in tissue infected with microbes that induce different classes of immune responses and various levels of pathology will be a powerful tool to define the factors controlling the fate of Treg cells.
References
Koch, M.A. et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 1–7 (2009).
Oldenhove, G. et al. Decrease of Foxp3(+) Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786 (2009).
Ivanov, I.I. et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349 (2008).
Hall, J.A. et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 29, 637–649 (2008).
Macpherson, A.J. & Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 4, 478–485 (2004).
Zheng, Y. et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356 (2009).
Chaudhry, A. et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991 (2009).
Lee, Y.K. et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009).
Xu, L., Kitani, A., Fuss, I. & Strober, W. Cutting edge: regulatory T cells induce CD4+CD25−Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 178, 6725–6729 (2007).
Yang, X.O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39 (2008).
Stock, P. et al. Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity. Nat. Immunol. 5, 1149–1156 (2004).
Sawitzki, B. et al. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J. Exp. Med. 201, 1925–1935 (2005).
Osorio, F. et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 38, 3274–3281 (2008).
Zhou, L. et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240 (2008).
Voo, K.S. et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 106, 4793–4798 (2009).
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007 (2009).
Wei, J. et al. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 104, 18169–18174 (2007).
Mantel, P.Y. et al. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 5, e329 (2007).
Hadjur, S. et al. IL4 blockade of inducible regulatory T cell differentiation: the role of Th2 cells, Gata3 and PU.1. Immunol. Lett. 122, 37–43 (2009).
Caretto, D., Katzman, S.D., Villarino, A.V., Gallo, E. & Abbas, A.K. Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J. Immunol. 184, 30–34.
Izcue, A. et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28, 559–570 (2008).
Curotto de Lafaille, M.A. et al. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126 (2008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Wohlfert, E., Belkaid, Y. Plasticity of Treg at infected sites. Mucosal Immunol 3, 213–215 (2010). https://doi.org/10.1038/mi.2010.11
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.11
This article is cited by
-
The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3+ regulatory T cells during mucosal inflammation
Mucosal Immunology (2019)
-
RORγt, a multitask nuclear receptor at mucosal surfaces
Mucosal Immunology (2017)
-
Immunity by equilibrium
Nature Reviews Immunology (2016)
-
Reciprocal interactions of the intestinal microbiota and immune system
Nature (2012)
-
Adaptive immunity in the host–microbiota dialog
Mucosal Immunology (2011)
