
Abstract
Celiac disease is a multifactorial disorder and provides a privileged model to decipher how the interplay between environmental and genetic factors can alter mucosal tolerance to a food antigen, lead to chronic intestinal inflammation, and ultimately promote T-cell lymphomagenesis. Here we summarize how HLA-DQ2/8 molecules, the main genetic risk factor for this disease can orchestrate a CD4+ T-cell adaptive immune response against gluten, and discuss recent data which shed light on the innate and adaptive immune stimuli that collaborate to induce a proinflammatory TH1 response, a massive expansion of intraepithelial lymphocytes, and a cytolytic attack of the epithelium. The intestinal immune response driven in genetically predisposed patients by chronic exposure to gluten emerges as the pathological counterpart of normal acute intestinal responses to intracellular pathogens.
Similar content being viewed by others

Introduction
Celiac disease (CD) likely appeared with one of the first environmental changes associated with human civilization, the development of agriculture 10,000 years ago in the Fertile Crescent, and the introduction of cereals in the human diet. The causal role of the diet was already considered in the first century AD by Arateus de Cappadoce, who provided the first description of the common manifestations, chronic diarrhea, abdominal distension, and progressive cachexia, and acknowledged their intestinal (coeliac) origin by naming the disease. But it was not until the 1950s that the triggering role of cereal storage proteins collectively called gluten was recognized by W Dicke, a young Dutch pediatrician who associated the onset of CD symptoms with the consumption of wheat bread and cereals and bestowed the first and still unique cure of the disease, the gluten-free diet.1
Many pieces of the puzzle of CD pathogenesis have since been progressively assembled. In 1957, the development of the Crosby capsule allowed M Shiner to examine duodenal biopsies and demonstrate the typical villous atrophy with hypertrophic crypts associated with this disease.2 Confirming the pioneering autopsy findings by S Gee in 1888, she simultaneously provided a rationale for the clinical symptoms of malnutrition and the first trusted diagnostic tool. This histological picture was completed in 1971 by A Ferguson who highlighted a hallmark of CD, the massive infiltration of the small intestinal epithelium by lymphocytes.3 In the early 1960s, familial studies underscored the contribution of predisposing genetic factors,4 the key role of which was established with the demonstration of a concordance rate of ∼75% in monozygotic twins vs. 20% in dizygotic twins and 10% in first degree relatives.5 In the 1970s, the observation of circulating immunoglobulin-G (IgG) and IgA antibodies against gluten and an autoantigen later identified as tissue transglutaminase II (Ttgase)6 provided new diagnostic tools and the first evidence that CD is immunologically driven, a concept rapidly supported by the discovery that the major genetic predisposing factor is in the human leukocyte antigen (HLA) complex. The identification of the HLA-DQ2 heterodimer as the major predisposing factor7, 8 and of its role in the development of an intestinal inflammatory CD4+ T-cell response to gluten was the next and main milestone, establishing a decisive link between the triggering environmental factor and the major predisposing gene.9 However, the puzzle remains incomplete. It is still unclear why only a subset of individuals with at risk HLA develop the disease, and why some do so very early in infancy closely after their first exposure to gluten, whereas others develop CD much later in adulthood. Furthermore, recent epidemiological studies have revealed the high prevalence (∼0.5–1%) of CD in the Western world and its wide clinical spectrum, raising new questions regarding the additional genetic and environmental factors that control the onset and severity of the disease, as well as the mechanisms that drive extra-digestive symptoms and complications. Among severe complications are autoimmune diseases, which affect approximately 20% of adult CD patients,10 underpinning the link between CD and autoimmunity, and malignancies, in particular rare but typical T-cell lymphomas. These lymphomas develop as a consequence of uncontrolled activation of intraepithelial lymphocytes (IELs), and provoke a severe enteropathy refractory to the gluten-free diet.11, 12 and 13 Here we summarize how HLA molecules can orchestrate an adaptive immune response to gluten in the intestinal mucosa and discuss recent observations that shed light on the auxiliary mechanisms necessary to trigger and/or perpetuate intestinal inflammation in CD.
HLA-DQ and Gluten Toxicity: How The Interplay Between Gluten and HLA-DQ2/8 Orchestrates an Adaptive CD4+ T-Cell Response in The Small Intestinal Lamina Propria of CD Patients
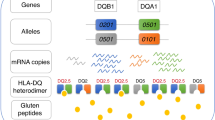
It is currently estimated that HLA genes account for ∼40% of the genetic predisposition in CD, and that susceptibility is primarily associated with specific MHC class II alleles (Figure 1). Thus, approximately 90–95% of CD patients express HLA-DQ2 heterodimers formed by an α-chain encoded by HLA-DQA1*05 and a β-chain encoded by HLA-DQB1*02 (either *0201 or *202). The genes can be inherited in cis (when present on one parental chromosome) as in the DR17 (formerly DR3) haplotype, or in trans (when the α- and β-chains are encoded by chromosomes from each parent) as in the DR7/DR11-13 (formerly DR5) haplotype. A very small number of patients may express exclusively DQB1*0202 or HLA-DQA1*05. The remaining 5–10% patients express the HLA-DQ8 heterodimer formed by an α- and a β-chain encoded by HLA DQA1*0301 and HLA-DQB1*0302, respectively. HLA-DQ2 and HLA-DQ8 share peptide binding motifs characterized by a preference of hydrophobic and negatively charged amino acids at specific positions (reviewed in references14, 15). The derivation of HLA-DQ2/8-restricted gluten specific CD4+ T-cell clones from the duodenal lamina propria of CD patients by L Sollid16 and subsequently by F Koning17 was the first evidence that these HLA molecules conferred CD susceptibility by promoting the presentation of gluten peptides to the intestinal adaptive immune system. Refined analysis has allowed investigators to decipher how this interplay is favored by the structural characteristics of gluten.

A keystone mechanism in celiac disease (CD) pathogenesis: the lamina propria adaptive CD4+ T-cell response to gluten orchestrated by HLA-DQ2/8 molecules. In active CD, gluten peptides left undigested by luminal and brush border enzymes can enter into the intestinal mucosa. Due to their primary sequence rich in Q-X-P motifs, gluten peptides are preferential substrates for tissue transglutaminase II (Ttgase). This enzyme is activated by tissue damage, and can deamidate neutral glutamine residues into negatively charged glutamic acid (left box). Negative charges in gluten peptides, as well as the presence of proline residues at specific positions, facilitate their binding into the peptide pocket of HLA-DQ2 (or -DQ8) expressed by antigen-presenting cells (APCs; including likely CD11c+ dendritic cells,33 and CD123+ plasmacytoid dendritic cells126; right box). Gluten presentation promotes the activation of a gliadin-specific TH1 CD4+ response in the intestinal lamina propria. Interferon (IFN)-γ can participate in the induction of mucosal damage.35
Since the pioneering work of W Dicke, cereal proteins responsible for CD have been extensively characterized. Harmful molecules, collectively called gluten, are the grain storage proteins contained in wheat, barley, and rye, that are unusually rich in proline (∼15%) and glutamine (∼30%), hence they are named prolamines. Toxicity of related proteins in oats remains debated. Wheat prolamines, the best characterized, comprise several hundred proteins divided in α- γ- and ω-gliadins (monomeric) and low and high MW glutenins. Both gliadins and glutenins contain intrachain disulfide bonds and show poor aqueous solubility. In addition, glutenins are extensively cross-linked by interchain disulfide bonds resulting in the formation of large protein aggregates further stabilized by hydrogen bonding between glutamine-rich repeats.18, 19 These characteristics, useful for bread making, underlie their toxicity for CD patients.
A first property is their unusual resistance to gastrointestinal enzymes. The hypothesis of a missing peptidase was first suggested by J Frazer in the early 1960s.20 To date, there is no evidence of a predisposing genetic defect, but Khosla and co-workers have demonstrated that the lack of endo-prolyl-peptidase activity in gastric and pancreatic enzymes and in the human brush border prevents efficient enzymatic attack of proline-rich domains in gluten proteins, allowing the release of large peptides that may reach the mucosal surface intact.18, 21 Notably, these authors showed that recombinant α2-gliadin submitted in vitro to conditions mimicking intraluminal digestion released a potent immunostimulatory 33 mer peptide consisting of a concatemer of several gluten T-cell epitopes.21, 22 More recently, the same authors drew similarities with prion-forming proteins where glutamine-rich repeats comparable to those found in prolamines can promote aggregation and impart proteolytic resistance.18
The second property imparted to prolamines by repeats rich in proline and glutamine is its ability to serve as a privileged substrate to Ttgase. This multifunction enzyme is constitutively expressed in the intestinal lamina propria and transiently activated upon tissue damage.23 It plays a physiological role in tissue repair by promoting protein cross-linking via the formation of isopeptide bonds between lysine and glutamine residues.24 It has been suggested that Ttgase cross-linked to gluten can be recognized as a hapten and stimulate the production of specific IgA. This attractive hypothesis to explain the appearance of CD-specific autoantibodies, however, could not be demonstrated even though the disappearance of autoantibodies occurs with a gluten-free diet.25 A second enzymatic activity of Ttgase is to deamidate neutral glutamine into negatively charged glutamic acid, an activity particularly efficient on glutamine-X-proline motifs frequent in prolamines26 (Figure 1). Introduction by Ttgase of negative charges in gluten peptides was shown to promote their interactions with positively charged residues in the peptide pocket of HLA-DQ2/8, resulting in stable HLA complexes that can be efficiently recognized by T cells.27, 28 (Figure 1). Deamidation is thus likely a crucial event in the generation of a full-blown gluten-specific T-cell response in CD. Notably, proline residues hamper peptide binding to most MHC class II molecules but on the contrary promote binding of gluten epitopes to HLA-DQ2/8 heterodimers, providing a complementary rationale for the preferential association of CD with HLA-DQ2/8.29, 30 and 31 Many potential T-cell epitopes susceptible to modification by Ttgase exist in α- and γ-gliadins, in glutenins, and in related prolamines, a dozen of which, mainly present in α- and γ-gliadins, were proven to evoke CD4+ T-cell responses in CD patients.32 The later epitopes are often clustered within large peptides resistant to intraluminal digestion such as the 33 mer endowed with enhanced immunostimulatory potency.32 Gluten-specific CD4+ T-cell lines and clones derived from CD patients were shown to produce interferon (IFN), notably in response to activated dendritic cells isolated from the mucosa of active CD.33 Consistent with a gliadin-driven TH1 response, IFN is found in large excess in the intestinal mucosa in active CD34 and may therefore participate to the severe mucosal damage.35
These results establish the central role of adaptive immunity in CD at the interface between the two main environmental and genetic factors. Its importance is further stressed by the evidence of an HLA-DQ2 gene dose effect. Thus, the risk of developing CD is fivefold higher in HLA-DQ2 homozygous then heterozygous individuals. This observation was correlated with the capacity of antigen-presenting cells (APCs) from homozygous individuals to elicit stronger gluten-specific T-cell responses than APCs from heterozygous subjects, a property ascribed to the higher density HLA-DQ2 molecules at their surface.36 Interestingly, the frequency of HLA-DQ2 homozygosity is increased in severe CD cases that become refractory to a gluten-free diet (to approximately 65 vs. 40% in uncomplicated CD)37, 38 and, in contrast, decreased (17%) in a small subset of patients with proven CD in childhood who spontaneously returned to latency with no symptoms and a normal histology after years of a diet containing substantial amounts of gluten.39 Similar to latent cases of CD that are revealed by screening CD family members and characterized by the development of intestinal lesions and symptoms later in life,40 several patients with spontaneous remissions had persistently elevated titers of anti-Ttgase antibodies and increased numbers of IELs. Moreover, two patients subsequently relapsed, indicating that tolerance to gluten remains fragile in these individuals.39
Why is Oral Tolerance to Gluten Broken in CD?
Previous studies have shown that, in mice with CD4+ T cells expressing a T-cell receptor specific for a dietary antigen, feeding with this antigen does not induce any enteropathy but rather results in the induction of regulatory T cells and tolerance associated with local production of antiinflammatory cytokines, interleukin (IL)-10 and transforming growth factor (TGF)-β (41 and accompanying review by C Berin and L Mayer). Accordingly, mice engineered to express HLA-DQ8 and a human CD4 molecule generated strong CD4+ T-cell responses when fed with gluten but did not develop enteropathy and their gluten-responsive T cells produced mainly TGF-β and IL-10, and no INF.42 Interestingly, crossing HLA-DQ8 mice on a NOD background resulted in the appearance of anti-Ttgase autoantibodies and of a skin disease reminiscent of herpetiformis dermatitis.43 In humans, this gluten-sensitive condition is generally associated with intestinal villous atrophy. However, T cells are absent in the skin lesions which only contain IgA deposits against a locally produced Ttgase and neutrophils, pleading against a T-cell-mediated disorder (44 and see below). Interestingly, HLA-DQ8×NOD mice did not develop any enteropathy, suggesting that the susceptibility to autoimmunity conferred by the NOD background was sufficient to promote an adaptive immune response and the production of autoantibodies but not sufficient to break T-cell tolerance in the gut.43 These data raise the question of the complementary mechanisms that contribute to breaking mucosal tolerance to gluten in CD, turning a controlled immune response into chronic inflammation and epithelial destruction. First clues were derived from observations unsatisfactorily explained by the anti-gliadin CD4+ T-cell response, namely the activation of IELs and the toxic effect that gliadin peptides not recognized by CD4+ T cells can exert in organ cultures on the intestinal mucosa of CD patients.
How The Interplay Between Adaptive and Innate Immune Mechanisms Orchestrated by IL-15 May Drive Iel Activation and Epithelial Damage
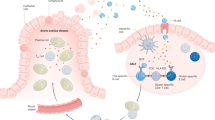
IELs have progressively emerged as important players in the pathogenesis of CD. Increased numbers of IELs can be observed in other small intestinal diseases notably in tropical sprue, giardiasis, Crohn's disease or autoimmune enteropathy, yet the massive increase in IELs with numbers often exceeding 70–80 IEL per 100 duodenal epithelial cells (<30/100 epithelial cells in controls) is a hallmark of active CD that subsides only partially after beginning a gluten-free diet (Figure 2).3 Furthermore, distinct subsets of IELs may exert opposite effects on the induction of the chronic epithelial lesions and their uncontrolled activation can lead to the most severe complication of CD, enteropathy-associated T-cell lymphomas.

The hypothetical interplay between innate and adaptive immune mechanisms leading to intraepithelial lymphocyte (IEL) activation and epithelial damage in celiac disease. In controls and in latent celiac disease (CD), CD94/NKG2A+ TCRγδ+ IEL may inhibit HLA-E+ CD8+ TCRαβ+ IELs via the secretion of transforming growth factor (TGF)-β (upper panel, left part). In active CD, the release of interleukin (IL)-15 by the epithelium may impair the inhibition exerted by TCRγδ+ IELs via TGF-β, enhance stimulatory signals delivered via the T-cell receptor, upregulate natural killer (NK) receptors (CD94, NKG2D) and/or their ligands (MIC), stimulate the effector functions of IELs and thereby promote epithelial damage (upper panel, right part). CD8+ TCRαβ IELs can be activated via their T-cell antigen receptor (TCR) by cross-presented gliadin peptides (lower panel, left box) or by other locally present antigens. The switch of CD94 from inhibitory to activatory functions and the upregulation of other activating NKR (NKG2D) and of their ligands on epithelium provide signals, which promote IELs activation via the TCR (lower panel, middle box). In addition, in active CD, interactions between NKR and their epithelial ligands become sufficient, in the presence of IL-15, to activate cytotoxicity and/or interferon (IFN)-γ secretion by TCRαβ+ IELs, and drive an autoimmune-like attack of the epithelium (lower panel, right box). The later mechanism is operating in refractory celiac sprue (RCS) where abnormal IELs lacking a TCR can exert their cytotoxicity against epithelial cells.
IEL subsets in CD
In humans, IELs in the normal small intestinal epithelium consist of approximately 75% CD8+ TCRαβ+ and 15% TCRγδ+ cells (mainly Vδ1+, either CD8+ or more often CD4−CD8−),45 and both subsets expand in active CD.46 Furthermore, these IELs in CD are enriched in cytolytic proteins (perforin, granzymes, FASL)47, 48 and 49 and produce large amounts of IFN-γ,50 indicating their likely contribution to the prominent apoptosis observed in the flattened surface epithelium.47, 48 After starting a gluten-free diet and parallel to villous architecture recovery, numbers of CD8+ TCRαβ+ IELs return to normal. In contrast, TCRγδ+ IELs remain increased for years, with percentages as high as 30–50% in many patients.46, 51 Moreover high numbers of TCRγδ+ IELs are found in patients with latent CD.39, 40 These observations led to the view, recently substantiated by experimental data, that CD8+ TCRαβ+ IELs exert a deleterious effect on epithelial cells that can be antagonized by TCRγδ+ IELs; and that this regulatory function of TCRγδ+ IELs is overwhelmed in active CD.51
Notably, in a small subset of CD patients who become refractory to a gluten-free diet, a third population of abnormal IELs expands massively whereas both CD8+ TCRαβ+ and TCRγδ+ IELs progressively disappear (notably TCRγδ+ before CD8+ TCRαβ+ IELs). These IELs possess a characteristic phenotype (CD103+, lacking surface CD3–TCR complexes but with intracellular CD3ɛ), clonal in- or out-of-frame rearrangements of the TcRγ chain(s), and chromosomal abnormalities, the later pointing to their malignant nature.11, 12, 52 This condition, called clonal refractory celiac sprue (or type II RCS), is considered a low-grade intraepithelial lymphoma53 and has a very poor prognosis, leading to intractable ulcerative duodenojejunitis, gastrointestinal, and extraintestinal dissemination of the abnormal IELs, and to their frequent transformation into a high-grade invasive T lymphoma usually also associated with a severe enteropathy54, 55 (Figure 3).

Interleukin (IL)-15 and T lymphomagenesis in celiac disease. In active celiac disease (CD), IL-15 is produced in excess by enterocytes and exerts potent antiapoptotic effects that prevent the elimination of activated intraepithelial lymphocytes (IELs) and promote their massive accumulation despite very low in situ proliferation. Chromosomal instability, demonstrated in active CD and likely due to inflammation (reviewed in reference160) may favor the emergence of malignant clonal IELs containing chromosomal abnormalities including a recurrent 1q trisomy. These IELs are highly sensitive to IL-15 and due to the presence of IL-15 can survive and escape elimination by safeguard proapoptotic mechanisms activated by DNA breaks. Abnormal IELs accumulate in the epithelium whereas normal IELs disappear progressively, defining clonal refractory celiac sprue (RCS) or type II RCS, now considered as a low-grade T-cell lymphoma. With time, some cells may acquire new mutations, which drive their transformation into high-grade T lymphoma likely independent of IL-15. The mechanisms underlying the emergence of abnormal IELs with a stereotypical phenotype (CD103+ lacking surface CD3–TCR complexes) in RCS remain unclear. In particular, it is unknown why these IELs which contain all CD3 subunits do not express a T-cell antigen receptor (TCR), even if in a few cases, functional clonal rearrangements of both the γ- and δ-chains have been detected at the molecular level (unpublished results).
Gluten-specific activation of CD8+TcRαβ+ IEL in active CD
Specific recognition of gluten peptides by CD8+ TCRαβ IELs is suggested by their exclusive expansion in active CD.46 Gianfrani and co-workers have recently observed that A-gliadin peptide 123–132 could be cross-presented to HLA class I restricted CD8+ TcRαβ+ cells in the intestinal mucosa of HLA-A2 CD patients56 and that CTL lines derived from the mucosa of the latter patients released granzyme B and IFN-γ when incubated with the peptide and an HLA-A2 epithelial cell line.57 Furthermore, adding the peptide to organ cultures from HLA-A2 but not from non-HLA-A2 CD patients induced a significant increase in CD25 and FASL on mucosal CD8+ cells, an intraepithelial migration of CD3+ cells and a concomitant increase in epithelial apoptosis, supporting the in vivo importance of this mechanism.57 A CD8+ adaptive response to gluten perhaps facilitated by the HLA-DQ driven CD4+ T-cell response may thus contribute to epithelial damage.
IL-15 driven activation of IELs in CD and RCS
Attention to IL-15 in CD was initially attracted by its capacity to stimulate the expression of two natural killer (NK) receptors (CD94 and NKG2D) on human IELs, both of which are upregulated on IELs in active CD (58, 59 and 60 and see below). IL-15 protein was subsequently found to be upregulated in enterocytes and lamina propria mononuclear cells in active CD and RCS.61, 62 IL-15 is a cytokine structurally related to IL-2 which differs, however, strikingly by its wide distribution—it can be produced by many cell types—as well as by its in vivo effects. In this regard, IL-15 is largely directed in mice toward NK, CD8 T cells, and CD8αα IELs, driving their differentiation, functional maturation, and survival (reviewed in references63, 64). In active CD and RCS, enterocyte-derived IL-15 may activate IELs by two possible mechanisms: (1) as a soluble cytokine binding the trimeric IL-15 receptor comprised by the βγ-module common with the IL-2 receptor and a specific α-chain (IL-15Rα) upregulated on IELs from active CD,61 and more likely, (2) as a cytokine bound to the ubiquitous RIL-15α chain at the surface of enterocytes, a complex that can very efficiently activate signal transduction in lymphocytes via the autonomous module.62, 65
IL-15 serves several functions in the epithelial compartment of CD patients. First, due to its potent antiapoptotic effect observed at low concentrations (10 times less than those necessary to induce proliferation), IL-15 stimulates the survival of IELs in uncomplicated CD and even more efficiently than that of clonal malignant IELs in RCS, providing a rationale for their massive accumulation despite a very modest increase in their proliferation rate.61, 62 In keeping with observations in IL-15 transgenic mice which develop CD8/NK lymphomas and leukemias,66 in RCS IL-15 may thereby prevent the elimination of transformed lymphocytes and be central in the emergence of T-cell lymphomas from IELs (Figure 3). Second, IL-15 has emerged as a driver of epithelial damage (Figure 2). Ex vivo, IL-15 stimulates effector functions of IELs from either active CD and RCS patients, notably their secretion of IFN-γ and their granzyme/perforin-dependent cytotoxicity against enterocyte lines.61, 62 In addition, neutralizing anti-IL-15 antibody inhibits the production of IFN-γ and epithelial apoptosis in organ cultures from active CD attesting to the important contribution of IL-15 to mucosal damage.67, 68 and 69 Furthermore, IL-15 may promote the activation of class I-restricted gliadin-specific IELs. Thus, in mice, IL-15 is necessary for the generation of high avidity specific cytotoxic T cells.70 Yet, IL-15 can also stimulate spontaneous killing of enterocyte lines by IELs from active CD as well as from RCS, the latter lacking a T-cell receptor, pointing to the contribution of additional receptors.61, 62 Recent work indicates that IL-15 may function in this regard by orchestrating the interactions between NK receptors on IELs and their ligands on epithelial cells.
NK receptor-driven activation of IELs in CD
A significant proportion of normal CD3+ IELs coexpress NK receptors and more particularly members of the lectin-like family, notably CD94 present in 30% normal IELs59 and NKG2D, expressed in humans not only by NK cells but also by the majority of CD8+ TcRαβ+ and TcRγδ+ lymphocytes including IELs (Figure 2).71 Findings in CD illustrate how these two receptors may finely tune human IELs activation.
NKG2D is an activating NK receptor associated in IELs with the intracellular adaptor molecule DAP10, which recruits the cytolytic machinery via activation of PI3K, ERK, and JNK kinases.58, 60 NKG2D responds to several ligands induced under conditions of stress which is thought to allow NK and cytotoxic T cells to track danger signals resulting from tissue infection or transformation.72 Strikingly, NKG2D and DAP10 are markedly upregulated in CD8 TCRαβ+ IELs from active CD, and epithelial expression of one NKG2D ligand, the MHC Ib molecule MICA, is strongly enhanced in both active CD and RCS.58, 60 Both changes might be driven by IL-15, which stimulates NKG2D and DAP10 expressions in cultured control IELs60 and epithelial MICA expression in intestinal organ cultures.58 NKG2D–MICA interactions can then turn on a cytolytic attack of the epithelium. In CD8 TCRαβ+ IELs, NKG2D acts as a coreceptor for the TCR, lowering its activation threshold.58, 71 NKG2D might thus promote the activation of MHC-class I gliadin-restricted CD8+ T cells, and perhaps reveal latent (low affinity) cross-reactivity to self antigens (Figure 2). Due to the presence of IL-15, the cytolytic attack of the epithelium may then be amplified and perpetuated via NKG2D independently of the TCR. Thus, NKG2D alone is sufficient to drive killing of MIC+ targets by CD8+TCRαβ+ IELs recently stimulated via their TCR, and subsequently cultured in the presence of IL-15.60 Accordingly, NKG2D activated TCR-independent killing of MIC+ targets by CD8+TCRαβ+ IELs from active CD60 (Figure 2). Notably, RCS IELs (which have no T-cell receptor) could also provoke NKG2D-mediated enterocyte killing,58 further suggesting that this pathway can somehow become independent of T-cell antigen receptor (TCR) stimulation in CD and result in a chronic autoimmune-like destruction of epithelium. Finally, a contribution of TCRγδ+ IELs to epithelial destruction in active CD is suggested by previous work showing that MIC+ epithelial targets can be killed by a subset of tumor-infiltrating V1 TCRγδ+ activated via both T-cell antigen receptor (TCR) and NKG2D dependent signals.73
A distinct and more complex scenario underlies the contribution of CD94 to IELs activation. The function of CD94 as an NK receptor is dictated by its association with NKG2 molecules within heterodimers that can bind the same ligand, HLA-E, another nonclassical MHC Ib molecule. CD94 can serve as an inhibitory receptor when associated with NKG2A, an adaptor molecule that recruits phosphatases via an ITIM motif present in its cytoplasmic tail. Upon binding to HLA-E, CD94/NKG2A delivers a negative signal that impairs the cytotoxicity of NK cells and negatively modulates T-cell receptor activation in memory CD8+ TcRαβ and TcRαα lymphocytes (reviewed in reference74). Notably, this heterodimer is expressed by the majority of CD94+ IELs in normal intestine, where it is acquired as a consequence of TCR stimulation and exerts a negative feed-back loop restraining TCR-mediated activation of CD8+ TCRαβ+ IELs.59, 75 A recent study of CD further suggests that a subset of TCRγδ+ IELs expressing both NKG2A and CD8 exerts a negative regulatory function on CD8+ TCRαβ+ IELs, inhibiting their production of IFN-γ and of the cytolytic protein granzyme in response to IL-15.51 This regulatory effect appeared mediated at least partially by TGF-β and was elicited either by TCR or NKG2A stimulation. As HLA-E was found to be upregulated on CD8+ TCRαβ+ IELs in active CD, the authors concluded that direct interactions between TCRγδ+ and CD8+ TCRαβ+ IELs51 (Figure 2) exist. These data, which need to be confirmed, however, provide an attractive rationale for the increase in TCRγδ IELs+ in patients with latent CD,39, 40, 46 and in keeping with data in mice suggesting that TCRγδ+ IELs exert a protective effect against epithelial damage induced by parasitic infections.76 In active CD, this mechanism may be overwhelmed due to the massive expression of IL-15, which can block the effect of TGF-β67 and to the reduced expression of NKG2A in both TCRγδ+ and CD8+ TCRαβ+ IELs51 (Figure 2).
Indeed, although the proportion of CD94+ CD8+ TcRαβ+ IELs is markedly increased in active CD, the vast majority does not express NKG2A, suggesting that this putative negative control imposed on IELs in the normal situation is removed in CD.59, 77 On the contrary, in active CD, CD94 appears to associate on a substantial fraction of IELs with a distinct NKG2 isotype, NKG2C that confers an activating function to the receptor.77 Indeed NKG2C binds the adaptor molecule DAP12 which, via an ITAM motif, recruits a signaling cascade that promotes cell proliferation, IFN-γ secretion and cytotoxicity independently of any other signals.74 Accordingly, in vitro engagement of NKG2C on CD8+ TcRαβ IEL from active CD stimulated their production of IFN, their proliferation and their cytotoxicity independently of any T-cell receptor engagement.77 As HLA-E is strongly upregulated on enterocytes in active CD, either as a consequence of the local secretion of IFN59, 77 or perhaps due to their stabilization on the cell surface by a subset of gliadin peptides (78 and see below), CD94/NKG2C IELs can likely initiate and perpetuate an autoimmune-like attack of epithelium in CD77 (Figure 2).
The mechanisms switching expression of CD94/NKG2A toward CD94/NKG2C heterodimers in CD have not been delineated. Although IL-15 can upregulate CD94 mRNA transcription, it does not seem able to modulate only NKG2A or NKG2C expression.79 Recent reports in mice provide however some hints concerning the mechanisms that may control expression of NK receptors in the intestine. Interestingly, NKG2A can be down-modulated by retinoic acid.80 This derivative of vitamin A, selectively produced by a subset of intestinal dendritic cells and likely also by enterocytes, plays an emerging key role in intestinal immune responses, controlling the homing of T cells and IgA cells, as well as the differentiation of regulatory T cells (reviewed in reference81). This additional effect of retinoic acid may be useful to promote the cytotoxic properties of IELs. Conversely, NKG2A is induced on CD8 T cells upon simultaneous stimulation by the T-cell receptor and TGF-β.82 This cytokine is a potent inhibitor of lymphocyte activation and intestinal inflammation able in particular to control lymphocyte proliferation and survival, IFN production and T-cell cytotoxicity (reviewed in reference83). Upregulation of NKG2A that possesses in its promoter a site for Smad3,82 a transcription factor central for the antiinflammatory effect of TGF-β,84 may participate to the down-modulating effect of TGF-β on cytotoxic T cells. In fact, TGF-β likely plays a more general role in the control of NK receptor expression. First, this cytokine downregulates NKG2D.85 Furthermore, in mice with T cells deprived of TGF-β receptor II, there is a massive expansion of highly cytotoxic T cells that develop an NK-like program with high levels of CD94, NKG2, DAP12, and NKG2D parallel to the onset of generalized inflammation and fatal autoimmunity.86 Interestingly, study of the transcriptome of human NKG2C+ IELs clones derived from active CD patients showed significant upregulation of several genes belonging to the NK cluster on chromosome 19p13, suggesting that NK reprogramming might be a feature of CD8+ TcRαβ+ IELs in CD.77 Our recent observation that IL-15 blocks the Smad3 pathway of TGF-β signaling in IELs and lamina propria lymphocytes of CD suggests that this cytokine may participate in the NK reprogramming of cytotoxic CD8+ T IELs.67 As in mice defective in TGF-β signaling, this reprogramming likely requires signals from the T-cell receptor, a hypothesis supported by the skewed T-cell repertoire of NKG2C+ IEL.77 The respective contribution of stimuli derived from gluten or from autoantigens remains to be delineated. Finally, appreciation of the role of the other NK receptors upregulated on CD IELs (NKP46, NKP44, NKP30)77 will await identification of their ligands.
In conclusion, these data indicate that IELs activated by IL-15 are a major cause of mucosal damage in CD. Mechanisms of IELs activation delineated in CD might result from the perversion of those normally requested to fight against intracellular pathogens and eliminate infected epithelial cells. Indeed IL-15 is induced by a variety of intracellular pathogens and was recently shown in mice to orchestrate the transient small intestinal enteropathy induced by intraperitoneal injection of poly-IC used to mimic stimulation of Toll-like receptor 3 by double-stranded RNA viruses.87 The rapid arrest of IL-15 synthesis after eviction of the pathogen avoids protracted epithelial damage. By contrast, in CD, the chronic upregulation of IL-15 results in durable epithelial damage, massive IEL hyperplasia and ultimately lymphomas. A question which remains to be solved concerns the mechanism driving IL-15 overproduction in CD patients. Recent controversial data suggest that gluten peptides might provide “innate”-like signals that contribute to triggering mucosal damage and IL-15 production independently of their capacity to bind HLA-DQ and activate adaptive immunity.
HLA-DQ Independent Toxicity of Gluten: A Stimulatory Effect of Gluten Peptides on Innate Immunity?
In the 1970s, organ culture was the first experimental approach to evaluate the toxicity of gluten-derived peptides. Gluten digests were shown to induce severe epithelial damage, and notably extensive apoptosis in intestinal biopsies from CD patients on a gluten-free diet, but not from controls.88, 89 and 90 Using this technique, D Kasarda showed that the N-terminus of A-gliadins contained several toxic fragments released by chymotrypsin digestion, including a large 31–55 fragment (p31–55).91 Toxicity was also observed in vivo after duodenal instillation of p31–5592 and in organ culture with the shorter p31–43 peptide.93 This result appeared quite paradoxical when neither p31–55 nor p31–43 proved usual CD4+ T-cell epitopes. Maiuri et al.94 were the first to suggest that the latter peptide might induce an innate immune response in CD. Notably, they observed that p31–43 induced a rapid production of IL-15 by lamina propria macrophages and/or dendritic cells, which in turn promoted the local CD4+ adaptive T-cell response, and stimulated IEL migration and epithelial apoptosis, all effects blocked by an anti-IL-15 antibody.94 These results were particularly interesting as they provided one possible mechanism to explain the massive upregulation of IL-15 in the intestinal mucosa of patients with active CD.59, 61, 62 Consistent with the possible role of p31–43 in the induction of IL-15 in the intestine of CD patients, we observed that adding a larger version of this peptide (p31–49) to organ culture from CD patients on a gluten-free diet stimulated epithelial expression of MICA, the target for NKG2D-mediated cytotoxicity of IELs in active CD (see above) and that this effect was blocked by an anti-IL-15 antibody.58 Yet, the persistent upregulation of IL-15 in the intestinal mucosa of patients who become refractory to a strict diet, suggests that complementary mechanisms underlie the induction of IL-15.62 Furthermore, the mechanism(s) through which p31—43/49 might induce IL-15 remain(s) elusive. Two groups observed that p31–43 induced a signaling cascade in organ culture of CD patients as well as in several cell types, resulting in tyrosine phosphorylation and actin rearrangement in epithelial cells95 and more generally mimicking or potentiating the effect of EGF receptor stimulation.96 Yet no specific receptor for p31–43 has yet been identified which may help substantiate this role. In addition, so far we have not been able to reproduce in vitro signaling by p31–49 in several enterocyte lines. Therefore, the exact role of p31–43/49 in the induction of IL-15 remains to be delineated.
Subsequent to observations that p31–43 induced the activation and/or maturation of dendritic cells in organ cultures from CD patients, several groups have observed that gliadin digests or peptides might stimulate the production of proinflammatory cytokines and/or the maturation of macrophages and/or dendritic cells of either murine and human origins.97, 98, 99, 100 and 101 Most studies took care to eliminate artifacts related to lipopolysaccharide contamination. Yet to date, the number and nature of peptides able to induce such effects and the involved mechanisms remain unclear, hampering efforts to delineate their exact contribution to CD pathogenesis.97, 98, 99, 100 and 101
Finally, an interesting recent study suggests that gliadin peptides may promote the expression of the nonclassical MHC molecule HLA-E at the surface of dendritic cells.78 This effect was ascribed to the homology between some α- and ϖ-derived gliadin peptides with amino-acid sequences known to bind the groove of HLA-E. Their binding stabilized HLA-E, resulting in enhanced surface expression. The authors observed that HLA-E expression protected dendritic cells against killing by NK cells expressing the HLA-E ligand CD94/NKG2A+, improving their dialogue with T cells and the stimulation of IFN.78 This mechanism may take place in the intestinal lamina propria of patients and promote the adaptive response. It is not excluded that this mechanism, together with IFN-γ, may contribute to the stimulation of HLA-E expression in the epithelial compartment, and thereby regulate IELs activation (see above).
A Role For The Humoral Response In CD Pathogenesis?
Whether IgA antibodies against either gluten or the autoantigen Ttgase are byproducts of the intestinal adaptive immune response or play a direct role in CD pathogenesis has remained uncertain, in as much as CD is particularly more frequent in patients with IgA deficiency. Nevertheless, the latter patients develop IgG antibodies, which may presumably play a comparable role.
IgA anti-Ttgase antibodies from active CD patients were reported to inhibit TGFβ-induced differentiation of the T84 enterocyte line in a three-dimensional gel,102 and more recently to disturb endothelial sprouting and migration of endothelial and vascular mesenchymal cells in an in vitro model of angiogenesis.103 An intriguing report indicates the presence in the sera of patients with active CD of antibodies cross-reactive with Ttgase, the VP7 protein of rotavirus, and several self-proteins including Toll-like receptor 4. Besides suggesting possible mimicry driven by rotavirus infection, the authors observed that these antibodies could enhance paracellular permeability of a T84 monolayer and activate monocytes.104 Yet, of note is the fact that deposits of IgA against Ttgase are present not only in active CD but also in the intestine of patients with latent CD without villous atrophy,105, 106 pleading against a major direct role of anti-Ttgase antibodies in intestinal epithelial damage. A recent report also suggests that human IgA and IgG against Ttgase may inhibit its activity in vitro as well as in situ.107 Yet, this finding is difficult to reconcile with the local deamidation of gluten-derived peptides which permits their binding to HLA-DQ2/8 molecules and their presentation to CD4+ lamina propria T cells. Thus, it is tempting to conclude that intestinal damage in CD is primarily driven by T cells whereas antibodies have only a minor direct contribution. Two sets of observations however suggest a possible role of antibodies in CD pathogenesis.
The first one concerns the extra-digestive manifestations of CD, more particularly dermatitis herpetiformis, where the papillary dermis is not infiltrated by T cells, but contains typical granular deposits of IgA directed against the local epidermal Ttgase III.44 Furthermore, IgA often associated with the presence of C3, was colocalized with epidermal Ttgase III in the vessel walls and within the scattered papillary peri- and intervascular granules. These results suggest that precipitation of immune complexes in the vessel walls may be an important pathogenic mechanism of dermatitis herpetiformis.108 The mechanisms by which gluten exposure promotes the production of antibodies against epidermal Ttgase III remain however elusive.
A second very recent observation concerns the capacity of IgA immune complexes to promote the retrotransport of intact gliadin peptides across the intestinal epithelium in patients with active CD (Figure 4). The role of a defective epithelial barrier that might promote the entrance of gluten peptides and the activation of the immune system has been hypothesized for a long time. Notably, the implication of the paracellular pathway has been considered. Indeed, intestinal paracellular permeability evaluated by electrical resistance and mannitol fluxes is increased in patients with active CD, and Fasano and co-workers observed that these parameters might be directly impaired by applying gliadin peptides to the luminal surface of human and mouse intestinal biopsies.109, 110 Furthermore, it was suggested that the polymorphism in the MYOX9B gene observed in a subset of CD patients may influence paracellular permeability. Yet this polymorphism is in a noncoding region and the exact role of MYOX9B, a nonconventional myosin, remains unclear.111

The retrotransport of immunoglobulin A (IgA)–gliadin complexes in active celiac disease. Gliadin peptides left undigested by luminal and brush border enzymes can be endocytosed. In controls, they can be further digested during their transepithelial transport likely due to their routing to lysosomes (left part). As a consequence, only very small amounts of intact peptides can reach the villous lamina propria. The entrance of gliadin peptides is however likely sufficient to drive a tolerogenic immune response and initiate the production of IgA in Peyer's patches and/or mesenteric lymph nodes. In the normal situation, gliadin peptides complexed to secretory IgA are likely entrapped in the mucus and eliminated by peristalsis (left part). In active celiac disease (CD), upregulation of CD71 and its abnormal expression at the apical surface of enterocytes allows the luminal capture of IgA–gliadin complexes which enter enterocytes with CD71 via the endosomal recycling pathway and can thus be rapidly delivered intact into the basolateral compartment (right part). The entrance of large amounts of intact gliadin peptides likely bolsters the local adaptive response. It is possible that IgA–gliadin complexes are particularly more efficient in driving the adaptive response and perhaps to help the switch from tolerance to inflammation, but this role remains to be demonstrated.
Data obtained by two groups, including ours, suggest on the contrary that gliadin peptides enter the mucosa via transcytosis and that this pathway is altered in active CD.112, 113 and 114 Using Ussing chambers to study the passage of radiolabeled or fluorescent peptides, it was shown that the immunostimulatory 33 mer (p56–89) enters by endocytosis in enterocyte lines,114 and that both the p31–49 peptide and/or the 33 mer were fully degraded during their passage across the intestinal mucosa of controls and patients on a gluten-free diet, likely due to their routing from endosomes toward the lysosomal pathway in enterocytes.112, 113 On the contrary, in active CD, not only was uptake of gliadin peptides increased,114 but, intact gliadin peptides were also rapidly translocated from the luminal surface to the serosal compartment.112, 113 There was however no evidence of nonspecific paracellular leakage of gliadin peptides,112 suggesting an abnormal protected pathway of transcytosis. By analogy with previous reports indicating that dietary antigens complexed to IgE or to IgG can be rapidly translocated into the lamina propria due to their respective binding to the low affinity IgE receptor CD23115 or to FcRn on epithelial cells,116 we hypothesized that gluten peptides may be complexed to intraluminal secretory IgA, bound to an IgA receptor and transported protected from lysosomal degradation by a specific transcytosis pathway.
We further implicated the CD71 receptor in transcytosis of IgA in CD113 (Figure 4). CD71 is well known for its role in the endocytosis of transferrin and is the prototype of the recycling receptors that protect their ligand from lysosomal degradation.117 This receptor was recently identified as a new receptor for polymeric IgA.118 Notably, CD71 is only expressed on the basolateral enterocyte membrane in the normal intestine and in patients on a gluten-free diet. In contrast, in active CD, CD71 expression is massively increased and CD71 is found at the apical enterocyte membrane where it colocalizes with IgA.113 This receptor efficiently binds polymeric and secretory, but not monomeric IgA. Consistent with its role in the retrotranscytosis of IgA–gliadin complexes, intestinal transport of intact gliadin peptides across the mucosa of patients with active CD was blocked by competition with irrelevant polymeric and secretory IgA, but not monomeric IgA, as well as by soluble CD71, but not CD89 (another IgA receptor not found on enterocytes)113 (Figure 4).
The contribution of IgA-mediated transcytosis of gliadin peptides to CD pathogenesis remains to be delineated. To be operative, it requires the presence of anti-gliadin IgA antibodies in the intestinal lumen, which may be induced as a normal protective adaptive response to gliadins. This is consistent with the thought that the luminal delivery of secretory IgA against food antigens and toxins is a protective mechanism to block these antigens from entering the body (Figure 4). This normal protective mechanism might be perverted in CD by the abnormal apical expression of CD71 which results on the contrary in enhanced mucosal delivery of gliadin peptides complexed to IgA (Figure 4). Two mechanisms may promote CD71 upregulation on enterocytes, low iron stocks119 and increased cell renewal. Both factors may sustain retrotranscytosis of IgA–gliadin complexes in active CD and thereby perpetuate the inflammatory immune response. It is however perhaps not excluded that upregulation of CD71 following infection or due to reduced iron stocks (a situation observed in infants and young women, two elective ages for CD onset) may contribute to the initiation of mucosal damage. Indeed, it is tempting to hypothesize that gliadin–IgA complexes are particularly immunostimulatory, and could provide a danger signal that is able to tip the local anti-gliadin immune response from tolerance to inflammation. In patients with IgA deficiency, a comparable role may perhaps be carried out by IgG and the FcRn receptor that can translocate IgG–antigens complexes across the mucosa116 and efficiently promote their presentation to mucosal dendritic cells.120
Alterations of The Mucosal Cytokine Network And Loss of Tolerance To Gluten: A Mixed and Intermingled Role of Innate and Adaptive Cytokines
As discussed above, tolerance to dietary antigens is ascribed to the activation of regulatory T cells and to the local production of immunosuppressive cytokines, IL-10 and TGF-β, which prevent excessive TH1 or TH2 responses (Figure 5). In active CD, intestinal inflammation is associated with an intense TH1 response as indicated by a considerable increase in IFN-γ transcripts34, 62 and the in situ activation of STAT1.121 In situ and ex vivo studies indicate that both gluten-specific CD4+ lamina propria T cells and IELs contribute to this massive TH1 response (see above). Defining the mechanisms that counteract tolerance mechanisms and drive the proinflammatory TH1 response in CD has been the focus of numerous studies. Surprisingly, and in marked contrast with other inflammatory bowel intestinal diseases, IL-12p40 transcripts are not increased in CD pleading against a driving role of IL-12 or IL-23.34, 62 Several other cytokines are possible candidates to drive and/or amplify the TH1 response in CD. The first one is IFN-γ, a known inducer of TH1 responses during viral infections.122 Attention to this cytokine was initially attracted by the onset of overt CD in patients receiving IFN-γ for therapeutic purposes,123 an observation also made in several autoimmune diseases (reviewed in reference124). Monteleone et al.123 subsequently observed that IFN-α: (1) is upregulated in the mucosa of active CD, (2) strongly potentiates the production of IFN-γ induced by T-cell receptor stimulation in explant cultures of human fetal gut,125 and (3) is produced by a subset of plasmacytoid dendritic cells recruited in the mucosa of active CD.126 Finally, they observed that antibodies against IFN-α inhibit the increase in IFN-γ transcripts induced by gliadin digests in biopsies from treated CD, demonstrating the contribution of this cytokine to the anti-gliadin TH1 response.126 Comparable experiments using neutralizing antibodies to block the transcription of IFN-γ and/or of the TH1 master regulator T-bet in organ cultures from untreated CD patients pleads against a role for IL-18, another cytokine present in excess in the mucosa of active CD patients127 but point to the complementary role of two other cytokines, IL-15 and IL-21.68 Notably, expression of the latter cytokine was found markedly increased in biopsies of active, but not treated CD patients, and could be induced by gluten digests in biopsies from treated CD patients.68 In contrast to IL-15 produced by epithelial cells, macrophages and dendritic cells, IL-21 is produced exclusively by CD4+ T cells (Figure 5).

The hypothetical interplay between innate and adaptive cytokines in celiac disease (CD). The mechanisms which switch a normal tolerogenic immune response to a dietary protein, such as gluten, into a deleterious proinflammatory response remain unclear. Three cytokines upregulated in active CD, interferon (IFN)-α, interleukin (IL)-15, and IL-21 may act in concert to promote the proinflammatory TH1 response and stimulate the dialogue between lamina propria and intraepithelial lymphocytes that lead to severe epithelial damage. The stimuli able to drive their upregulation and their respective contribution are schematized. The inhibitory effect of IL-15 on transforming growth factor (TGF)-β signaling and of IL-21 and IL-15 on the response of effector T cells to FOXP3 regulatory T cells may perhaps promote the emergence of autoimmune complications if their upregulation can be driven outside the gut, which remains to be demonstrated.
The interplay between IFN-α and IL-15 produced by innate cells and/or enterocytes and IL-21 produced by CD4+ T cells may not only stimulate the intestinal TH1 response but serve other functions converging to break tolerance to gluten and promote the fatal dialogue between lamina propria CD4+ T cells and IELs that culminate into severe epithelial damage (Figure 5). On the one hand, IFN-α and IL-15 can stimulate each others synthesis and both are potent inducers of dendritic cell maturation.122, 128, 129 Moreover, in mice, IFN-γ can stimulate crosspriming of CD8+ T cells against soluble antigens,130 suggesting its possible role in the induction of MHC class I gliadin-restricted CD8+ T cells which could then be further activated and expanded by IL-15130 (Figure 5). On the other hand, the IL-21 signaling pathway partially overlaps with that of IL-15, the IL-21 receptor comprising a specific IL-21Rα chain, and the γc chain common with the IL-15 receptor.131 The two cytokines exert both opposing and synergistic effects at the interface between innate and adaptive immunity. In marked contrast with IL-15, IL-21 alone inhibits the maturation of dendritic cells131, 132 and downregulates the expression and activation of the NKG2D receptor in human NK and CD8+ T cells,133 both effects unexpected in active CD. It is however conceivable that these effects are overcome in the presence by IL-15 and/or IFN-α. In fact, IL-21, which alone has only moderate effects on CD8 T cells, exerts potent synergistic effects with IL-15 on enhancing CD8 T-cell survival, proliferation, production of IFN-γ and granzyme/perforin-dependent cytotoxicity.134 IL-21 produced by CD4+ T cells may therefore be one actor in the dialogue between lamina propria CD4+ T cells and IELs (Figure 5). IL-21 might also act in concert with IL-15 to break the local immunoregulatory mechanisms that negatively control TH1 responses. Thus, we have observed that IL-15 inhibits the Smad3 pathway of TGF-β signaling, a key regulatory pathway to avoid intestinal inflammation.67 This effect of IL-15, resulting from the activation of the JNK pathway, was observed not only in IELs but also in CD4+ lamina propria cells. In keeping with the uncontrolled TH1 response observed in mice with T cells deprived of TGF-β receptor II, IL-15-mediated blockade of Smad3 was shown to promote the production of IFN-γ in biopsies from untreated CD patients.67 Finally, IL-15 in concert with IL-21 might impair a second important regulatory pathway in the small intestine. In humans, FoxP3+ regulatory T cells play a major role in preventing not only autoreactivity in the small intestine but also adverse responses to food antigens.135 Recent data do not favor a genetic link of CD with the FOXP3 gene136 and the number of FoxP3+ cells detected in situ is increased in patients with either active or latent CD.137 However both IL-15 and IL-21 can block the suppressive effects of FoxP3+ CD4+CD25+ regulatory T cells on CD4+ effector T cells (138 and BenAhmed et al., submitted), suggesting that the latter cells, even if recruited may be ineffective to control the TH1 inflammatory response (Figure 5). The inhibitory effect of IL-15 on two important immunoregulatory mechanisms may perhaps participate in the induction of autoimmune diseases in CD, a notion supported by the analysis of IL-15 transgenic mice which develop multiorgan lymphocytic infiltrates and aggressive skin lesions.66
These results raise the question of the mechanisms and signaling pathways, which promote the excessive production of IFN-α, IL-15, and IL-21 in CD. Recent studies point to the role of complementary genetic and environmental factors.
The Contribution of Additional Genetic Factors: A Role For The IL-2/IL-21 Region?
As over 60% of familial clustering remains unexplained by HLA genes, the identification of additional genetic loci appears an important chance to gain new insight into CD pathogenesis. The contribution of other genes in the HLA region (TNF-α, LT-α, MICA) has been suggested but results are inconsistent and conclusions hampered by the very strong linkage disequilibrium in this region. Linkage analyses have pointed to several candidate regions among which 5q31–33 and 19p13.1, the later containing MYO9B and ICAM-1 which have emerged as candidate genes from further studies (111, 139 and reviewed in reference140). Yet these findings have been inconsistently replicated. A candidate gene approach, recently confirmed by a genome wide association analysis, suggests a link with the CTLA-4 region (2q33) in some populations.140, 141 Yet, the functional consequence of the observed polymorphism remains unknown.
Very recently, genome-wide association studies based on the use of single nucleotide polymorphisms have shed new light on the genetics of complex polygenic diseases. These analyses support the concept that the majority of genetic variation contributing to disease susceptibility is carried by multiple variants of weak size effects.140 Two recent studies led in CD by van Heel and co-workers on a total of roughly 3,400 patients with CD and 4,800 controls of UK, Irish, and Dutch origins have revealed a robust association with eight distinct new chromosomal regions.142, 143 Yet, the newly identified variants were estimated to account for only 3–4% of the genetic susceptibility of CD, indicating that many other true associations remain undetected. Furthermore, the alleles that were more frequent in CD cases and conferred either protective or risk effects with weak odds ratios between 0.7 and 1.4 for all detected variants.140, 142, 143
How these genes contribute to CD pathogenesis will await identification of the causal variants. Interestingly, however, seven out of the eight identified regions harbored genes with known immune functions, three of which (4q27, 3p21 and 12p24) have been already associated with other autoimmune diseases, particularly type I diabetes, whereas, a fourth one (2q11–2q12) has been associated with Crohn's disease.140, 142, 143 Possession of these variants might influence the quality or intensity of the immune responses providing perhaps a selective advantage to resist to infectious agents, which may explain their selection during evolution in many individuals. Reciprocally, they might lower the threshold of immune reactivity, allowing for inappropriate reactions against autoantigens or harmless environmental antigens. Along this line, it is interesting that the most significant association outside the HLA region was observed with single nucleotide polymorphisms located in 4q27 in a block of four genes including IL2 and IL21,142, 143 an association recently confirmed in a Norwegian cohort.144 Due to extensive linkage disequilibrium, the causal variant associated with CD could not be identified. Yet, it is noticeable that 4q27 is syntenic to the idd3 region on murine chromosome 3 that controls the development of type I diabetes in NOD mice as well as the sensitivity to several experimental autoimmune diseases.145 No precise mutation was identified in mice but the susceptibility idd3 haplotype was associated with reduced IL-2 transcription in response to T-cell stimulation, and a lower proportion and impaired function of CD4+CD25+ regulatory T cells. In contrast, no change in the transcription of IL-21 was observed.145 In humans, the possible impact of this association on IL-2 or IL-21 transcription is unknown. It is tempting to suggest an impact on IL-21 because, as indicated above, CD4 lamina propria T cells from patients of active CD produce increased amounts of IL-21 which seems important to promote the CD intestinal TH1 response. Yet, increased IL-21 expression is observed in a large variety of TH-1 type gastrointestinal inflammatory processes,146 further pleading for the idea that the polymorphisms associated with inflammatory/autoimmune diseases might exert their effect by finely tuning common immune pathways.
Finally, it is interesting to observe that the second strongest association outside the HLA region detected by the studies by van Heel and co-workers is within a region specifically associated with CD and in 1q31,142, 143 a region encompassed by the recurrent 1q trisomy characteristic of RCS malignant IELs.147 Notably, this region contains RGS1, a gene encoding a negative regulator of G-protein which is specifically expressed in IELs.142, 143 No association has however been detected so far with regions encoding IL-15 or IFN-α or their receptors.
The Contribution of Additional Environmental Factors: A Role In The Induction of IFN-α and IL-15?
The ability of gastrointestinal infections as a trigger to overt CD is a long lasting hypothesis. The first suggested mechanism was molecular mimicry between gluten and proteins from pathogens such as the EIB protein of adenovirus type 12148 or the hyphal wall protein 1 from Candida albicans,149 but this hypothesis was not supported by experimental evidence. Furthermore, the presence of rod-shaped bacteria adhering to the duodenal mucosa of CD has been noted,150 yet their adherence may be secondary to local changes in the glycocalyx and evidence of their direct role in disease triggering is lacking.
In contrast, that IFN-α and of IL-15 are important players in CD pathogenesis as well as key actors of the early innate responses to intracellular pathogens point to the role of viral infections, notably by double-stranded RNA viruses which, via TLR3 or intracellular sensors, are strong inducers of both cytokines.87, 122, 129 This hypothesis is supported by a recent epidemiological survey indicating that repeated infections by rotavirus, a double-stranded RNA virus, promote the onset of childhood CD.151 It is therefore conceivable that induction of IFN-α and IL-15 as a consequence of viral infections can reinstruct previously tolerogenic dendritic cells and/or stimulate the de novo recruitment and activation of proinflammatory dendritic cells which can in turn prime gluten specific T cells and tip the local immune balance from tolerance to inflammation. Interestingly, in mice, stimulation of TLR-3 by a peritoneal injection of poly-IC was shown to trigger a transient enteropathy. One study showed that tissue damage switched on the deamidation activity of Ttgase 2, a mechanism which may stimulate gluten loading of HLA-DQ molecules on dendritic cells and subsequently may amplify priming of gluten-specific T cells.23 Another study demonstrated the rapid induction of IL-15 in enterocytes and of NKG2D on IELs and showed that blockade of IL-15 and NKG2D could inhibit epithelial damage,87 suggesting that IELs can also be rapidly activated on TLR3 stimulation. The mechanisms which maintain IFN-α and IL-15 synthesis long after eviction of the putative pathogen(s) and which reactivate their production depending on patients exposure to gluten remain however to be deciphered. Notably, the respective contribution of so-called innate and adaptive gluten peptides needs to be better defined as well as the signaling pathways that can unlock IL-15 translation.
From Cognitive Studies to Therapy?
Knowledge on the pathogenesis of CD has rapidly progressed and CD is now the first HLA-associated disease where the role of HLA has been elucidated. Lessons from CD might thus help to decipher pathogenic mechanisms of other HLA-associated autoimmune diseases, several of which can be associated with CD and/or share genetic and environmental risk factors. Another outcome of this cognitive work is to provide rationales to improve diagnostic tools and elaborate new preventive or therapeutic strategies.
On the basis of previous epidemiological studies, a preventive strategy in infants with at risk HLA is currently being tested which consists in the introduction of small amounts of gluten in 4-month-old babies “protected” by breast-feeding with the hope of preserving/promoting tolerance to gluten.152 The triggering role of repeated rotavirus infections, if confirmed, may become an additional justification for vaccination. The identification and/or generation of “nontoxic” but good bread-making wheat breeds is an attractive proposal to prevent CD.124 It can be feared however that the later approach remains the quest of the Holy Grail given the complexity of wheat genetics, the abundance of immunogenic sequences and because the structural characteristics of gluten proteins that confer immunogenicity seem largely overlapping with those that underlie their bread-making qualities.
As the gluten-free diet is a real burden for many patients, several alternatives to a lifelong diet are considered, including the use of immunomodulatory vaccines, of Ttgase inhibitors,153 HLA-DQ blockers,154 or exogenous endoprolyl peptidases (21 and reviewed in reference155). Developing a treatment as perfectly safe and effective as the gluten-free diet is however a difficult challenge. To date, the safest and most promising approach appears the use of novel highly efficient exogenous prolyl endoproteases,156, 157 as a similar strategy has been used for many years to treat pancreatic insufficiency. Taken with the food, these enzymes may help patients to deal with occasional lapses in their diet or may protect highly sensitive individuals from inadvertent presence of gluten in food products. Nevertheless, the efficiency of this approach still needs precise assessment. Bethune and co-workers have recently taken advantage of the identification of a small population of macaques which develop a gluten-sensitive enteropathy158 to test this strategy. Administration of the enzyme during gluten challenge prevented the clinical relapse but not the reappearance of antibodies against gliadin and Ttgase,159 leading to question the long-term safety.
The need of an alternative strategy is more particularly indispensable for the small subsets of patients who develop clonal RCS, become resistant to dietary changes and have a very poor prognosis despite all treatments attempted so far.38 Blocking IL-15 or its signaling pathway may help to control epithelial damage and above all to induce the apoptosis of abnormal IELs and prevent their expansion and further transformation into high-grade lymphomas.
Conclusion
It is now much clearer how one of the first environmental changes introduced by human civilization has resulted in the emergence of a disease in individuals with a predisposed immune system. The keystone is the adaptive immune response elicited by the interplay between at risk HLA and gluten. Several other pieces of the puzzle underlying CD pathogenesis have been picked up, which underscore the complementary role of innate immune mechanisms. Yet, their assemblage remains hypothetical and some pieces are still controversial or lacking, notably to explain the diversity in the age at onset and in the presentation. Handy and pertinent animal models are strongly needed to authenticate the respective role of each putative player, establish their influence and hierarchy in the cascades that lead to mucosal damage, and ultimately define relatable tools to test innovative therapeutic strategies.
Disclosure
The authors declared no conflict of interest.
References
Van de Kamer, J., Weijers, H. & Dicke, W. Coeliac disease V. Some experiments on the cause of the harmful effect of wheat gliadin. Acta Paediatr. Scand. 42, 223–231 (1953).
Sakula, J. & Shiner, M. Coeliac disease with atrophy of the small-intestine mucosa. Lancet 273, 876–877 (1957).
Ferguson, A. & Murray, D. Quantitation of intraepithelial lymphocytes in human jejunum. Gut 12, 988–994 (1971).
Carter, C., Sheldon, W. & Walker, C. The inheritance of coeliac disease. Ann. Hum. Genet. 23, 266–278 (1959).
Greco, L. et al. The first large population based twin study of coeliac disease. Gut 50, 624–628 (2002).
Dieterich, W. et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 3, 797–801 (1997).
Howell, M.D., Austin, R.K., Kelleher, D., Nepom, G.T. & Kagnoff, M.F. An HLA-D region restriction fragment length polymorphism associated with celiac disease. J. Exp. Med. 164, 333–338 (1986).
Sollid, L.M., Markussen, G., Ek, J., Gjerde, H., Vardtal, F. & Thorsby, E. Evidence for a primary association of celiac disease to a particular HLA-DQ a/b heterodimer. J. Exp. Med. 169, 345–350 (1989).
Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 18, 53–81 (2000).
Cosnes, J. et al. Incidence of autoimmune diseases in celiac disease: protective effect of the gluten-free diet. Clin. Gastroenterol. Hepatol. 6, 753–758 (2008).
Cellier, C. et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group [see comments]. Lancet 356, 203–208 (2000).
Cellier, C. et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology 114, 471–481 (1998).
Spencer, J. et al. Enteropathy-associated T cell lymphoma (malignant histiocytosis of the intestine) is recognized by a monoclonal antibody (HML-1) that defines a membrane molecule on human mucosal lymphocytes. Am. J. Pathol. 132, 1–5 (1988).
Sollid, L. Coeliac disease: dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 9, 647–655 (2002).
Kagnoff, M.F. Celiac disease: pathogenesis of a model immunogenetic disease. J. Clin. Invest. 117, 41–49 (2007).
Lundin, K.E.A. et al. Gliadin-specific, HLA-DQ(a1*0501,b1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J. Exp. Med. 178, 187–196 (1993).
Van de Wal, Y. et al. Small intestinal T cells of celiac disease patients recognize a natural pepsin fragment of gliadin. Proc. Natl. Acad. Sci. USA 95, 10050–10054 (1998).
Bethune, M.T. & Khosla, C. Parallels between pathogens and gluten peptides in celiac sprue. PLoS Pathog. 4, e34 (2008).
Wieser, H. Chemistry of gluten proteins. Food Microbiol. 24, 115–119 (2007).
Frazer, A. et al. Gluten-induced enteropathy: the effect of partially digested gluten. Lancet 2, 252–255 (1959).
Shan, L. et al. Structural basis for gluten intolerance in celiac sprue. Science 297, 2218–2220 (2002).
Qiao, S.W. et al. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J. Immunol. 173, 1757–1762 (2004).
Siegel, M. et al. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE 3, e1861 (2008).
Fesus, L. & Piacentini, M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem. Sci. 27, 534–539 (2002).
Sollid, L.M., Molberg, O., McAdam, S. & Lundin, K.E. Autoantibodies in coeliac disease: tissue transglutaminase—guilt by association? Gut 41, 851–852 (1997).
Vader, L. et al. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J. Exp. Med. 195, 643–649 (2002).
Molberg, O. et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 4, 713–717 (1998).
Van de Wal, Y. et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specfic T cell reactivity. J. Immunol. 161, 1585–1588 (1998).
Kim, C.Y., Quarsten, H., Bergseng, E., Khosla, C. & Sollid, L.M. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. USA 101, 4175–4179 (2004).
Qiao, S.W. et al. Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J. Immunol. 175, 254–261 (2005).
Stepniak, D. et al. Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J. Immunol. 180, 3268–3278 (2008).
Vader, L.W. et al. Characterization of cereal toxicity for celiac disease patients based on protein homology in grains. Gastroenterology 125, 1105–1113 (2003).
Raki, M. et al. A unique dendritic cell subset accumulates in the celiac lesion and efficiently activates gluten-reactive T cells. Gastroenterology 131, 428–438 (2006).
Nilsen, E., Lundin, K., Krajci, P., Sollid, L. & Brantzaeg, P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with TH1 or TH0 profile dominated by interferon g. Gut 37, 766–776 (1995).
Guy-Grand, D., Disanto, J.P., Henchoz, P., Malassis-Seris, M. & Vassalli, P. Small bowel enteropathy—role of intraepithelial lymphocytes and of cytokines (IL-12, IFN-g, TNF) in the induction of epithelial cell death and renewal. Eur. J. Immunol. 28, 730–744 (1998).
Vader, W. et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc. Natl. Acad. Sci. USA 100, 12390–12395 (2003).
Al-Toma, A. et al. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin. Gastroenterol. Hepatol. 3, 315–319 (2006).
Malamut, G. et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I and type II. Gastroenterology (in press).
Matysiak-Budnik, T. et al. Long-term follow-up of 61 coeliac patients diagnosed in childhood: evolution toward latency is possible on a normal diet. Gut 56, 1379–1386 (2007).
Mäki, M., Holm, K., Collin, P. & Savilahti, E. Increase in g/d T cell receptor bearing lymphocytes in normal small bowel mucosa in latent celiac disease. Gut 32, 1412–1414 (1991).
Weiner, H. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 182, 207–214 (2001).
Black, K., Murray, J. & David, C.S. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J. Immunol. 169, 5595–5600 (2002).
Marietta, E. et al. A new model for dermatitis herpetiformis that uses HLA-DQ8 transgenic NOD mice. J. Clin. Invest. 114, 1090–1097 (2004).
Sardy, M., Karpati, S., Merkl, B., Paulsson, M. & Smyth, N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J. Exp. Med. 195, 747–757 (2002).
Jarry, A., Cerf-Bensussan, N., Brousse, N., Selz, F. & Guy-Grand, D. Subsets of CD3+(TCRab or TCRgd) and CD3− lymphocytes isolated from normal human gut epithelium differ from their PBL counterparts. Eur. J. Immunol. 20, 1097–1103 (1990).
Kutlu, T. et al. Numbers of T cell receptor (TCR) a/b+ but not of TCR g/d+ intraepithelial lymphocytes correlate with the grade of villous atrophy in coeliac patients on a long term normal diet. Gut 34, 208–214 (1993).
Ciccocioppo, R. et al. Increased enterocyte apoptosis and Fas-Fas ligand system in celiac disease. Am. J. Clin. Pathol. 115, 494–503 (2001).
Di Sabatino, A. et al. Intraepithelial and lamina propria lymphocytes show distinct patterns of apoptosis whereas both populations are active in Fas based cytotoxicity in coeliac disease. Gut 49, 380–386 (2001).
Oberhuber, G. et al. Evidence that intestinal intraepithelial lymphocytes are activated cytotoxic T cells in celiac disease but not in Giardiasis. Am. J. Pathol. 148, 1351–1357 (1996).
Olaussen, R.W. et al. Interferon-gamma-secreting T cells localize to the epithelium in coeliac disease. Scand. J. Immunol. 56, 652–664 (2002).
Bhagat, G. et al. Small intestinal CD8TCRgammadeltaNKG2A intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J. Clin. Invest. 118, 281–293 (2008).
Verkarre, V. et al. Recurrent partial trisomy 1q22-q42 in clonal intraepithelial lymphocytes in refractory celiac sprue. Gastroenterology 125, 40–46 (2003).
Badgi, E., Diss, T., Munson, P. & Isaacson, P. Mucosal intraepithelial lymphocytes in enteropathy-associated T-cell lymphoma, unlcerative jejunitis, and refractory celiac sprue constitute a neoplastic population. Blood 94, 260–264 (1999).
Daum, S., Cellier, C. & Mulder, C.J. Refractory coeliac disease. Best Pract. Res. Clin. Gastroenterol. 19, 413–424 (2005).
Malamut, G. et al. Adult celiac disease with severe or partial villous atrophy: a comparative study. Gastroenterol. Clin. Biol. 32, 236–242 (2008).
Gianfrani, C. et al. Celiac disease association with CD8(+) T cell responses: identification of a novel gliadin-derived HLA-A2-restricted epitope. J. Immunol. 170, 2719–2726 (2003).
Mazzarella, G. et al. Gliadin activates HLA class I-restricted CD8+ T cells in celiac disease intestinal mucosa and induces the enterocyte apoptosis. Gastroenterology 134, 1017–1027 (2008).
Hue, S. et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 21, 367–377 (2004).
Jabri, B. et al. Selective expansion of intraepithelial lymphocytes expressing the HLA-E-specific natural killer receptor CD94 in celiac disease. Gastroenterology 118, 867–879 (2000).
Meresse, B. et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366 (2004).
Di Sabatino, A. et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 55, 469–477 (2006).
Mention, J.J. et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 125, 730–745 (2003).
Budagian, V., Bulanova, E., Paus, R. & Bulfone-Paus, S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 17, 259–280 (2006).
Fehniger, T.A. & Caligiuri, M.A. Interleukin 15: biology and relevance to human disease. Blood 97, 14–32 (2001).
Bergamaschi, C. et al. Intracellular interaction of IL-15 with its receptor alpha during production leads to mutual stabilization and increased bioactivity. J. Biol. Chem. (2007).
Fehniger, T. et al. Fatal leukemia in interleukin15 transgenic mice follows early expansion in natural killer and memory phenotype CD8+ T cells. J. Exp. Med. 193, 219–231 (2001).
Ben Ahmed, M. et al. Inhibition of TGF-beta signaling by interleukin-15 via phospho-c-jun upregulation: a new role for interleukin-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 132, 994–1008 (2007).
Fina, D. et al. Interleukin-21 contributes to the mucosal T helper cell type 1 response in celiac disease. Gut (2007).
Maiuri, L. et al. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology 119, 996–1006 (2000).
Oh, S., Perera, L.P., Burke, D.S., Waldmann, T.A. & Berzofsky, J.A. IL-15/IL-15Ralpha-mediated avidity maturation of memory CD8+ T cells. Proc. Natl. Acad. Sci. USA 101, 15154–15159 (2004).
Roberts, A.I. et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 167, 5527–5530 (2001).
Raulet, D. Roles of NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3, 781–790 (2003).
Groh, V., Steinle, A., Bauer, S. & Spies, T. Recognition of stress-induced MHC molecules by intestinal gammadelta T cells. Science 279, 1737–1740 (1998).
Jabri, B. & Meresse, B. NKG2 receptor-mediated regulation of effector CTL functions in the human tissue microenvironment. Curr. Top Microbiol. Immunol. 298, 139–156 (2006).
Jabri, B. et al. TCR specificity dictates CD94/NKG2A expression by human CTL. Immunity 17, 487–499 (2002).
Roberts, S.C. et al. T-cell ab+ and gd+ deficient mice display abnormal but distinct phenotypes toward a natural, widespread infection of the intestinal epithelium. Proc. Natl. Acad. Sci. USA 93, 11774–11779 (1996).
Meresse, B. et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J. Exp. Med. 203, 1343–1355 (2006).
Terrazzano, G. et al. Gliadin regulates the NK-dendritic cell cross-talk by HLA-E surface stabilization. J. Immunol. 179, 372–381 (2007).
Lieto, L.D., Borrego, F., You, C.H. & Coligan, J.E. Human CD94 gene expression: dual promoters differing in responsiveness to IL-2 or IL-15. J. Immunol. 171, 5277–5286 (2003).
Laouar, A. et al. Cutting edge: distinct NK receptor profiles are imprinted on CD8 T cells in the mucosa and periphery during the same antigen challenge: role of tissue-specific factors. J. Immunol. 178, 652–656 (2007).
Mora, J., Iwata, M. & von Andrian, U. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat. Rev. Immunol. (2008)[Epub ahead of print].
Gunturi, A., Berg, R.E., Crossley, E., Murray, S. & Forman, J. The role of TCR stimulation and TGF-beta in controlling the expression of CD94/NKG2A receptors on CD8 T cells. Eur. J. Immunol. 35, 766–775 (2005).
Rubtsov, Y.P. & Rudensky, A.Y. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 7, 443–453 (2007).
Yang, X. et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 18, 1280–1291 (1999).
Castriconi, R. et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 100, 4120–4125 (2003).
Marie, J.C., Liggitt, D. & Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25, 441–454 (2006).
Zhou, R., Wei, H., Sun, R., Zhang, J. & Tian, Z. NKG2D recognition mediates Toll-like receptor 3 signaling-induced breakdown of epithelial homeostasis in the small intestines of mice. Proc. Natl. Acad. Sci. USA 104, 7512–7515 (2007).
Falchuk, Z.M., Gebhard, R.L., Sessoms, C. & Strober, W. An in vitro model of gluten-sensitive enteropathy. Effect of gliadin on intestinal epithelial cells of patients with gluten-sensitive enteropathy in organ culture. J. Clin. Invest. 53, 487–500 (1974).
Falchuk, Z.M. et al. Gluten-sensitive enteropathy. Influence of histocompatibility type on gluten sensitivity in vitro. J. Clin. Invest. 66, 227–233 (1980).
Maiuri, L. et al. FAS engagement drives apoptosis of enterocytes of coeliac patients. Gut 48, 418–424 (2001).
de Ritis, G. et al. In vitro (organ culture) studies of the toxicity of specific A-gliadin peptides in celiac disease. Gastroenterology 94, 41–49 (1988).
Sturgess, R. et al. Wheat peptide challenge in coeliac disease. Lancet 343, 758–761 (1994).
Maiuri, L. et al. In vitro activities of A-gliadin-related synthetic peptides: damaging effect on the atrophic coeliac mucosa and activation of mucosal immune response in the treated coeliac mucosa. Scand. J. Gastroenterol. 31, 247–253 (1996).
Maiuri, L. et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362, 30–37 (2003).
Maiuri, L. et al. Unexpected role of surface transglutaminase type II in celiac disease. Gastroenterology 129, 1400–1413 (2005).
Barone, M.V. et al. Growth factor-like activity of gliadin, an alimentary protein: implications for celiac disease. Gut 56, 480–488 (2007).
Cinova, J. et al. Gliadin peptides activate blood monocytes from patients with celiac disease. J. Clin. Immunol. 27, 201–209 (2007).
De Stefano, D. et al. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J. Mol. Med. 84, 65–74 (2006).
Palova-Jelinkova, L. et al. Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J. Immunol. 175, 7038–7045 (2005).
Thomas, K.E., Sapone, A., Fasano, A. & Vogel, S.N. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J. Immunol. 176, 2512–2521 (2006).
Tuckova, L. et al. Activation of macrophages by gliadin fragments: isolation and characterization of active peptide. J. Leukoc. Biol. 71, 625–631 (2002).
Halttunen, T. & Maki, M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology 116, 566–572 (1999).
Myrsky, E. et al. Coeliac disease-specific autoantibodies targeted against transglutaminase 2 disturb angiogenesis. Clin. Exp. Immunol. 152, 111–119 (2008).
Zanoni, G. et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 3, e358 (2006).
Kaukinen, K. et al. Small-bowel mucosal transglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: a prospective and randomized clinical study. Scand. J. Gastroenterol. 40, 564–572 (2005).
Tosco, A. et al. Immunoglobulin A anti-tissue transglutaminase antibody deposits in the small intestinal mucosa of children with no villous atrophy. J. Pediatr. Gastroenterol. Nutr. 47, 293–298 (2008).
Caputo, I., Barone, M.V., Martucciello, S., Lepretti, M. & Esposito, C. Tissue transglutaminase in celiac disease: role of autoantibodies. Amino Acids (2008).
Preisz, K., Sardy, M., Horvath, A. & Karpati, S. Immunoglobulin, complement and epidermal transglutaminase deposition in the cutaneous vessels in dermatitis herpetiformis. J. Eur. Acad. Dermatol. Venereol. 19, 74–79 (2005).
Fasano, A. et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 355, 1518–1519 (2000).
Lammers, K.M. et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 135, 194–204 e3 (2008).
Monsuur, A.J. et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat. Genet. 37, 1341–1344 (2005).
Matysiak-Budnik, T. et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125, 696–707 (2003).
Matysiak-Budnik, T. et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 205, 143–154 (2008).
Schumann, M. et al. Mechanisms of epithelial translocation of the alpha(2)-gliadin-33mer in coeliac sprue. Gut 57, 747–754 (2008).
Yang, P., Berin, M.C., Yu, L., Conrad, D. & Perdue, M. Enhanced intestinal transepithelial antigen transport in allergic rats is mediated by IgE and CD23 (FcepsilonRII). J. Clin. Invest 106, 879–886 (2000).
Yoshida, M. et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20, 769–783 (2004).
Yamashiro, D.J., Tycko, B., Fluss, S.R. & Maxfield, F.R. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell 37, 789–800 (1984).
Moura, I.C. et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 194, 417–425 (2001).
Barisani, D. et al. Adaptive changes of duodenal iron transport proteins in celiac disease. Physiol. Genomics 17, 316–325 (2004).
Qiao, S.W. et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc. Natl. Acad. Sci. USA 105, 9337–9342 (2008).
Mazzarella, G. et al. Constitutive activation of the signal transducer and activator of transcription pathway in celiac disease lesions. Am. J. Pathol. 162, 1845–1855 (2003).
Biron, C.A. Interferons a and b as immune regulators-a new look. Immunity 14, 661–664 (2001).
Monteleone, G. et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut 48, 425–429 (2001).
Stepniak, D. & Koning, F. Celiac disease—sandwiched between innate and adaptive immunity. Hum. Immunol. 67, 460–468 (2006).
Monteleone, G., Pender, S.L.F., Wathen, N.C. & MacDonald, T.T. Interferon-a drives T-cell mediated immunopathology in the intestine. Eur. J. Immunol. 31, 2247–2255 (2001).
Di Sabatino, A. et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology 133, 1175–1187 (2007).
Salvati, V.M. et al. Interleukin 18 and associated markers of T helper cell type 1 activity in coeliac disease. Gut 50, 186–190 (2002).
Gary-Gouy, H., Lebon, P. & Dalloul, A.H. Type I interferon production by plasmacytoid dendritic cells and monocytes is triggered by viruses, but the level of production is controlled by distinct cytokines. J. Interferon Cytokine Res. 22, 653–659 (2002).
Mattei, F., Schiavoni, G., Belardelli, F. & Tough, D.F. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 167, 1179–1187 (2001).
Le Bon, A. et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 176, 4682–4689 (2006).
Leonard, W.J. & Spolski, R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat. Rev. Immunol. 5, 688–698 (2005).
Brandt, K., Singh, P.B., Bulfone-Paus, S. & Ruckert, R. Interleukin-21: a new modulator of immunity, infection, and cancer. Cytokine Growth Factor Rev. 18, 223–232 (2007).
Burgess, S.J., Marusina, A.I., Pathmanathan, I., Borrego, F. & Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 176, 1490–1497 (2006).
Zeng, R. et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 201, 139–148 (2005).
Torgerson, T.R. et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology 132, 1705–1717 (2007).
Bjornvold, M. et al. FOXP3 polymorphisms in type 1 diabetes and coeliac disease. J. Autoimmun. 27, 140–144 (2006).
Tiittanen, M., Westerholm-Ormio, M., Verkasalo, M., Savilahti, E. & Vaarala, O. Infiltration of forkhead box P3-expressing cells in small intestinal mucosa in coeliac disease but not in type 1 diabetes. Clin. Exp. Immunol. 152, 498–507 (2008).
Peluso, I. et al. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J. Immunol. 178, 732–739 (2007).
Abel, M. et al. Adulthood-onset celiac disease is associated with intercellular adhesion molecule-1 (ICAM-1) gene polymorphism. Hum. Immunol. 67, 612–617 (2006).
Dubois, P.C. & van Heel, D.A. Translational mini-review series on the immunogenetics of gut disease: immunogenetics of coeliac disease. Clin. Exp. Immunol. 153, 162–173 (2008).
Djilali-Saiah, I. et al. CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut 43, 187–189 (1998).
Hunt, K.A. et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. (2008).
van Heel, D.A. et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 39, 827–829 (2007).
Adamovic, S. et al. Association study of IL2/IL21 and FcgRIIa: significant association with the IL2/IL21 region in Scandinavian coeliac disease families. Genes Immun. 9, 364–367 (2008).
Yamanouchi, J. et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat. Genet. 39, 329–337 (2007).
Meresse, B., Verdier, J. & Cerf-Bensussan, N. The cytokine interleukin 21: a new player in coeliac disease? Gut 57, 879–881 (2008).
Verkarre, V. et al. Refractory celiac sprue is a diffuse gastrointestinal disease. Gut 52, 205–211 (2003).
Kagnoff, M.F., Austin, R.K., Hubert, J.J., Bernardin, J.E. & Kasarda, D.D. Possible role for a human adenovirus in the pathogenesis of celiac disease. J. Exp. Med. 160, 1544–1557 (1984).
Nieuwenhuizen, W.F., Pieters, R.H., Knippels, L.M., Jansen, M.C. & Koppelman, S.J. Is Candida albicans a trigger in the onset of coeliac disease? Lancet 361, 2152–2154 (2003).
Forsberg, G. et al. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am. J. Gastroenterol. 99, 894–904 (2004).
Stene, L.C. et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am. J. Gastroenterol. 101, 2333–2340 (2006).
Troncone, R., Ivarsson, A., Szajewska, H. & Mearin, M.L. Review article: future research on coeliac disease—a position report from the European multistakeholder platform on coeliac disease (CDEUSSA). Aliment Pharmacol. Ther. 27, 1030–1043 (2008).
Siegel, M., Xia, J. & Khosla, C. Structure-based design of alpha-amido aldehyde containing gluten peptide analogues as modulators of HLA-DQ2 and transglutaminase 2. Bioorg. Med. Chem. 15, 6253–6261 (2007).
Xia, J. et al. Cyclic and dimeric gluten peptide analogues inhibiting DQ2-mediated antigen presentation in celiac disease. Bioorg. Med. Chem. 15, 6565–6573 (2007).
Cerf-Bensussan, N., Matysiak-Budnik, T., Cellier, C. & Heyman, M. Oral proteases: a new approach to managing coeliac disease. Gut 56, 157–160 (2007).
Gass, J., Bethune, M.T., Siegel, M., Spencer, A. & Khosla, C. Combination enzyme therapy for gastric digestion of dietary gluten in patients with celiac sprue. Gastroenterology 133, 472–480 (2007).
Stepniak, D. et al. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G621–G629 (2006).
Bethune, M.T. et al. A non-human primate model for gluten sensitivity. PLoS ONE 3, e1614 (2008).
Bethune, M.T., Ribka, E., Khosla, C. & Sestak, K. Transepithelial transport and enzymatic detoxification of gluten in gluten-sensitive rhesus macaques. PLoS ONE 3, e1857 (2008).
Verkarre, V., Romana, S.P. & Cerf-Bensussan, N. Gluten-free diet, chromosomal abnormalities, and cancer risk in coeliac disease. J. Pediatr. Gastroenterol. Nutr. 38, 140–142 (2004).
Acknowledgements
INSERM U793 is supported by grants from INSERM, Université Paris Descartes, Agence Nationale pour la Recherche, Fondation pour la Recherche Médicale, Fondation Princesse Grace, Association Française des Intolérants au Gluten.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meresse, B., Ripoche, J., Heyman, M. et al. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol 2, 8–23 (2009). https://doi.org/10.1038/mi.2008.75
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2008.75
This article is cited by
-
The gliadin-CFTR connection: new perspectives for the treatment of celiac disease
Italian Journal of Pediatrics (2019)
-
T lymphocytes in the intestinal mucosa: defense and tolerance
Cellular & Molecular Immunology (2019)
-
Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis
Cell Death & Disease (2019)
-
Defective proteostasis in celiac disease as a new therapeutic target
Cell Death & Disease (2019)
-
Diversity of oat varieties in eliciting the early inflammatory events in celiac disease
European Journal of Nutrition (2014)
