
Abstract
Inflammatory bowel diseases (IBDs) are caused by an aberrant and excessive local immune response to components of the bacterial microflora that are: poorly controlled by endogenous counter regulatory mechanisms such as the immunosuppressive cytokine transforming growth factor-β1 (TGF-β1). Studies in human IBD tissues have documented a disruption of TGF-β1 signaling marked by a block in the phosphorylation of the activated TGF-β receptor-associated signaling molecule, Smad3, caused by the upregulation of the intracellular inhibitor of Smad signaling, Smad7. Inhibition of Smad7 with a specific antisense oligonucleotide restores TGF-β1/Smad3 signaling, resulting in a marked suppression of inflammatory cytokine production. The functional relevance of Smad7 in gut inflammation was confirmed by studies in murine models of IBD. In inflamed tissues of mice with colitis induced by either the trinitrobenzene sulfonic acid or oxazolone, p-Smad3 was low despite active TGF-β1 being produced in excess. In vivo administration of Smad7 antisense oligonucleotides to mice with colitis restored TGF-β1 signaling and decreased the synthesis of inflammatory molecules and the extent of gut damage. These data support a role for Smad7 in maintaining intestinal inflammation, and suggest that blocking Smad7 could be a promising way to dampen the ongoing inflammation in IBD.
Similar content being viewed by others


Introduction
Crohn's disease and ulcerative colitis, the major forms of inflammatory bowel disease (IBD) in humans, result from the interaction of genetic and environmental factors to ultimately promote an immunopathologic process leading to chronic inflammation in the gut.1 Recent insights into the nature of these diseases derived mainly from studies in experimental models of colitis suggest that IBD is caused by an aberrant local immune response to components of the bacterial microflora, inappropriately controlled by endogenous counter regulatory mechanisms.2 One of these mechanisms involves the production/activity of transforming growth factor-β1 (TGF-β1). TGF-β1 knockout mice also develop systemic inflammation involving the gut and die early in life.3 Animal models in which T cells cannot respond to TGF-β1 also develop wasting disease and gut inflammation.4 Moreover, many studies in murine models of IBD have shown that synthesis of TGF-β1 is associated with either complete protection from the development of colitis or diminished severity of colitis.5, 6, 7
Smad Signaling in IBD
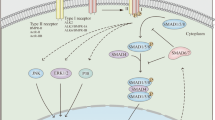
TGF-β1 initiates signaling through the ligand-dependent activation of a complex of heterodimeric transmembrane serine–threonine kinases consisting of type I (TGF-βRI) and type II (TGF-βRII) receptors. On TGF-β1 binding, there is phosphorylation and activation of TGF-βRI by the constitutively active and autophosphorylating TGF-βRII. TGF-βRI, in turn, phosphorylates two proteins, termed Smad2 and Smad3, for their high homology to the Drosophila Mad and the Caenorhabditis elegans Sma proteins. Once phosphorylated, Smad2 and Smad3 associate with Smad4 and translocate to the nucleus where Smad protein complexes participate in transcriptional control of target genes (Figure 1).8 Targeted disruption of Smad3 is associated with diminished cell responsiveness to TGF-β1. Mutant mice also exhibit massive infiltration of T cells and pyogenic abscess formation in the stomach and intestine, supporting the view that Smad3 is an essential mediator of the TGF-β1-induced anti-inflammatory and suppressive activities.9 Consistent with these observations, we have shown that active TGF-β1 is produced in the normal human gut and that both colonic mucosal and lamina propria mononuclear cell (LPMCs) samples express high levels of phosphorylated (p)-Smad3. It can be noted that the blockade of endogenous TGF-β1 activity in cultures of normal mucosal explants and LPMC results in enhanced synthesis of inflammatory cytokines, including Th1-type-associated markers (Table 1), thus indicating that TGF-β1 has a decisive function in negatively controlling inflammatory pathways in the human gut.10 By contrast, phosphorylation of Smad3 is defective in the inflamed gut of patients with Crohn's disease and patients with ulcerative colitis, even though TGF-β1 is expressed at high levels in these patients.11, 12 Moreover, in vitro studies with LPMC isolated from the colon of IBD patients revealed that these cells do not phosphorylate Smad3 and maintain high levels of inflammatory cytokines, following stimulation with exogenous TGF-β1.11

Binding of transforming growth factor-β1 (TGF-β1) to the type II receptor causes the activation of the type I receptor, which in turn phosphorylates Smad2/3. These proteins thus become able to interact with Smad4, and the complex Smad2/3/Smad4 migrates into the nucleus and binds to the DNA. (b) The inhibitor Smad7 interacts with TGF-β1 type I receptor and prevents Smad3 phosphorylation, thus blocking the TGF-β1-associated Smad signaling pathway illustrated in (a).
The Smad family also includes two inhibitory proteins, termed Smad6 and Smad7, that occupy the ligand-activated TGF-β1RI and interfere with the phosphorylation of Smad2/Smad3 (Figure 1). Upregulation of Smad6 and Smad7 is associated with inhibition of TGF-β1-induced Smad signaling.8, 13 Analysis of these two proteins in IBD tissues revealed that Smad7 is overexpressed in whole mucosal and LPMC samples, whereas Smad6 was barely detectable in both normal and IBD mucosa (11, and personal unpublished observations). It can be noted that specific antisense oligonucleotides for Smad7 reduced Smad7 protein in IBD LPMC, thereby making these cells responsive to exo-genous TGF-β1, as indicated by the enhanced phosphorylation of Smad3 and diminished expression of inflammatory cytokines. Consistently, in inflamed mucosal tissue explants from Crohn's disease patients, the inhibition of Smad7 restored Smad3 phosphorylation and decreased inflammatory cytokine production, an effect that was partially blocked by a neutralizing TGF-β1 antibody.11
We have extended these studies to examine the interactions between Smad signaling and nuclear factor (NF)-κB activation in IBD LPMC, as NF-κB is supposed to sustain intestinal inflammation.1, 2 Although TGF-β1-inhibited TNF-α-induced NF-κB activation in normal LPMC, it had no activity in LPMC from IBD patients. This was due to overexpression of Smad7, as treatment of IBD LPMC with antisense to Smad7 allowed TGF-β1 to rapidly downregulate NF-κB activation.14
High Smad7 expression and defective TGF-β-associated Smad3 activation also occur in the stomach of patients with Helicobacter pylori-associated gastritis.15 By contrast, Smad7 expression is not upregulated in the duodenum of patients with active celiac disease, where impaired TGF-β-Smad3-dependent transcriptional responses are caused by interleukin (IL)-15-driven c-jun-N-terminal kinase activation.16 Overall, these data indicate that Smad7 upregulation is not a specific hallmark of IBD and that its induction is not simply an epiphenomenon of the ongoing mucosal inflammation.
Regulation of Smad7 in IBD
As Smad7 appears to be important in the regulation of TGF-β1 in the gut, understanding how this inhibitor is regulated in IBD could help design new therapeutic interventions for patients. Experiments with cell lines have shown that Smad7 can be induced by activators of NF-κB (e.g., TNF-α and IL-1β) and STAT-1 (e.g., interferon-γ and IL-7). These transcription factors are hyperactivated in IBD, but Smad7 protein expression remained unchanged after blocking interferon-γ/STAT-1 or TNF-α/NF-κB-signaling pathways in IBD LPMC.11, 17 Smad7 is also rapidly induced by TGF-β1 itself, thus representing an important effector in the feedback loop that controls TGF-β1/Smad signaling.13 However, it seems unlikely that this mechanism is responsible for increasing Smad7 expression in IBD, as p-Smad3 is reduced in samples exhibiting high Smad7 levels.11
A detailed analysis of Smad7 RNA revealed no difference between IBD and normal intestinal samples, indicating that in IBD, Smad7 is regulated at the post-transcriptional level.18 Indeed, it was shown that Smad7 is ubiquitinated and targeted for proteasome degradation in control but not IBD tissue. This seems to be secondary to the different status of acetylation of Smad7, as acetylation and ubiquitination compete for the same lysine residues of the protein. So acetylation prevents ubiquitination and protects Smad7 protein against proteasomal degradation.17 Smad7 was highly acetylated in vivo in IBD but not control samples.18 We have shown that in IBD, the transcriptional coactivator p300 interacts with Smad7 and promotes its acetylation. Consistent with this, the inhibition of p300 by silencing diminished acetylation and expression of Smad7 in Crohn's disease LPMC,18 thus suggesting that manipulating the p300 level in IBD tissue can be useful for controlling TGF-β1 activity and eventually limiting the local inflammation.
Smad7 Controls Gut Inflammation in Murine Models of IBD
In accordance with data in humans, high levels of active TGF-β1 are seen in the inflamed tissues of mice with both trinitrobenzene sulfonic acid and oxazolone-mediated colitis.19 This is associated with reduced phosphorylation of Smad3 and high expression of Smad7. Oral administration of Smad7 antisense oligonucleotide reduced Smad7 and restored TGF-β1-associated p-Smad3 expression in the colon of mice with either trinitrobenezene sulfonic acid- or oxazolone colitis. Treatment with Smad7 antisense led to significant amelioration of both forms of colitis, as evidenced by a reduction in weight loss and macroscopic and microscopic evidence of inflammation.19 Smad7 antisense oligonucleotide also reduced inflammation in a model of chronic colitis induced by repeated administrations of trinitrobenezene sulfonic acid. Analysis of colonic cytokines revealed that restoration of TGF-β1 signaling by inhibition of Smad7 resulted in a significant downregulation of Th1-cytokines (IL-12 and interferon-γ) and reduced expression of Th1-associated transcription factors (i.e., T-bet and STAT-1) in trinitrobenezene sulfonic acid colitis. On the other hand, oral administration of Smad7 antisense oligonucleotide reduced the production of IL-4 in mice with oxazolone-induced colitis.19 These studies suggest that resolution of gut inflammation may be accomplished by downregulating Smad7 and allowing endogenous TGF-β1 to inhibit inflammatory pathways that promote tissue injury (Table 2). However, in this context, it can be noted: that TGF-β1 has different effects on different cell types and that inhibiting Smad7 in some cell types might enhance the detrimental effects of TGF-β1, such as the production of collagen and fibrosis.
Disclosure
Giovanni Monteleone has filed a patent titled “Antisense modulation of sMad7 expression.” He has also appeared as an expert witness for the US Department of Justice. Thomas T. MacDonald has received consulting fees from Danone and Abbott, lecture fees from Danone, and grant funding from GSK and Danone. He has also been an expert witness for the US Department of Justice.
References
Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 347, 417–429 (2002).
Bouma, G. & Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 3, 521–533 (2003).
Kulkarni, A.B. & Karlsson, S. Transforming growth factor-β1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol. 143, 3–9 (1993).
Goreli, L. & Flavell, R.A. Transforming growth factor-β in T-cell biology. Nat. Rev. Immunol. 2, 46–53 (2002).
Neurath, M.F. et al. Experimental granulomatous colitis in mice is abrogated by induction of TGF-β—mediated oral tolerance. J. Exp. Med. 183, 2605–2616 (1996).
Boirivant, M., Fuss, I.J., Chu, A. & Strober, W. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J. Exp. Med. 188, 1929–1939 (1998).
Powrie, F., Carlino, J., Leach, M.W., Mauze, S. & Coffman, R.L. A critical role for transforming growth factor-β but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 183, 2669–2674 (1996).
Heldin, C.-H., Kohei, M. & ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 (1997).
Yang, X. et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T-cell responsiveness to TGF-β. EMBO J. 18, 1280–1291 (1999).
Di Sabatino, A. et al. Blockade of transforming growth factor-β upregulates T-box transcription factor T-bet, and increases T-helper-cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut 57, 605–612 (2008).
Monteleone, G. et al. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Invest. 108, 601–609 (2001).
Babyatsky, M.W., Rossiter, G. & Podolsky, D.K. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 110, 975–984 (1996).
Nakao, A. et al. Identification of Smad7, a TGF-β-inducible antagonist of TGF-β signalling. Nature 389, 631–635 (1997).
Monteleone, G. et al. A failure of transforming growth factor-β1 negative regulation maintains sustained NF-κB activation in gut inflammation. J. Biol. Chem. 279, 3925–3932 (2004).
Monteleone, G. et al. Induction and regulation of Smad7 in the gastric mucosa of patients with Helicobacter pylori infection. Gastroenterology 126, 674–682 (2004).
Benahmed, M. et al. Inhibition of TGF-β signaling by IL-15, a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 132, 994–1008 (2007).
Monteleone, G., Pallone, F. & MacDonald, T.T. Smad7 in TGF-β-mediated negative regulation of gut inflammation. Trends Immunol. 25, 513–517 (2004).
Monteleone, G. et al. Post-transcriptional regulation of Smad7 in the gut of patients with inflammatory bowel disease. Gastroenterology 129, 1420–1429 (2005).
Boirivant, M. et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-β1-mediated suppression of colitis. Gastroenterology 131, 1786–1798 (2006).
Acknowledgements
The authors have received support for work on the function of Smad7 in the gut from the Fondazione “Umberto Di Mario,” Rome, the Broad Medical Research Program Foundation, and Giuliani, S.p.A, Milan, Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Monteleone, G., Boirivant, M., Pallone, F. et al. TGF-β1 and Smad7 in the regulation of IBD. Mucosal Immunol 1 (Suppl 1), S50–S53 (2008). https://doi.org/10.1038/mi.2008.55
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2008.55
This article is cited by
-
Immune Inhibitory Properties and Therapeutic Prospects of Transforming Growth Factor-Beta and Interleukin 10 in Autoimmune Hepatitis
Digestive Diseases and Sciences (2022)
-
High Smad7 in the early post-operative recurrence of Crohn’s disease
Journal of Translational Medicine (2020)
-
Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling
International Journal of Colorectal Disease (2020)
-
Current and emerging therapeutic targets for IBD
Nature Reviews Gastroenterology & Hepatology (2017)
-
Intraarticular overexpression of Smad7 ameliorates experimental arthritis
Scientific Reports (2016)
