
Abstract
ADMIRE was a multicenter, randomized-controlled, open, phase IIB superiority trial in previously untreated chronic lymphocytic leukemia. Conventional front-line therapy in fit patients is fludarabine, cyclophosphamide and rituximab (FCR). Initial evidence from non-randomized phase II trials suggested that the addition of mitoxantrone to FCR (FCM-R) improved remission rates. Two hundred and fifteen patients were recruited to assess the primary end point of complete remission (CR) rates according to International Workshop on Chronic Lymphocytic Leukemia criteria. Secondary end points were progression-free survival (PFS), overall survival (OS), overall response rate, minimal residual disease (MRD) negativity and safety. At final analysis, CR rates were 69.8 FCR vs 69.3% FCM-R (adjusted odds ratio (OR): 0.97; 95% confidence interval (CI): (0.53–1.79), P=0.932). MRD-negativity rates were 59.3 FCR vs 50.5% FCM-R (adjusted OR: 0.70; 95% CI: (0.39–1.26), P=0.231). During treatment, 60.0% (n=129) of participants received granulocyte colony-stimulating factor as secondary prophylaxis for neutropenia, a lower proportion on FCR compared with FCM-R (56.1 vs 63.9%). The toxicity of both regimens was acceptable. There are no significant differences between the treatment groups for PFS and OS. The trial demonstrated that the addition of mitoxantrone to FCR did not increase the depth of response. Oral FCR was well tolerated and resulted in impressive responses in terms of CR rates and MRD negativity compared with historical series with intravenous chemotherapy.
Similar content being viewed by others

Introduction
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative disorder accounting for 30% of adult leukemia and 25% of non-Hodgkin lymphoma. CLL is the commonest form of leukemia above the age of 50 years with a median age of diagnosis of 70 years. The treatment of CLL is tailored around the physical state of the patient owing to toxicity associated with the chemotherapy-based treatments.
CLL is still an incurable disease, and most patients will eventually become resistant to treatment. For physically fit patients, combination chemoimmunotherapy in the form of fludarabine, cyclophosphamide and rituximab (FCR) has become the standard of care based on evidence from large randomized controlled and non-randomized trials.1, 2, 3 Updated analysis suggested an improvement in progression-free survival (PFS) and overall survival (OS) in patients treated with FCR over FC.1 Hence, this combination is considered to be the gold-standard first-line treatment in patients deemed to be suitable for fludarabine-based treatment.
The addition of mitoxantrone to fludarabine-based therapy has been found to induce high response rates in a variety of lymphoproliferative disorders, including follicular non-Hodgkin lymphoma4 and mantle cell lymphoma.5 The addition of mitoxantrone to fludarabine and cyclophosphamide (FCM) has been assessed in a phase II clinical trial in which 69 CLL patients requiring therapy were given this combination as front-line treatment.6 This trial reported a complete remission (CR) rate of 64% with minimal residual disease (MRD)-negativity rate of 26% and overall response rate (ORR) of 90%. The same group reported the combination of FCM-R in 72 previously untreated patients resulting in an ORR of 93% and a CR rate of 82% of which 46% achieved an MRD-negative CR7 that appeared higher than expected for FCR. FCM-R has also been reported in patients with relapsed/refractory CLL. Two trials involving 60 and 29 patients with relapsed refractory CLL reported an ORR with FCM of 78% and 79%, respectively, with 30 (50%) and 9 (32%) patients, achieving a CR.8, 9 We previously reported a randomized phase II trial of 52 patients with relapsed CLL, with ORR with FCM and FCM-R of 58% and 65%, respectively,10 and an acceptable toxicity profile. Eight (15.4%) patients in this trial achieved MRD negativity.
The ADMIRE (Does the ADdition of Mitoxantrone Improve REsponse to FCR chemotherapy in patients with CLL?) trial was designed to assess whether the addition of mitoxantone to FCR increases the depth of response in previously untreated patients with CLL requiring therapy in comparison to the standard FCR treatment. The current literature suggests that patients who respond to therapy and do not have detectable CLL by extremely sensitive techniques have a significantly prolonged survival.11, 12, 13 MRD with a sensitivity of 10−4 has become an important end point in the treatment of CLL especially in the era of chemoimmunotherapy. Indeed, attainment of MRD negativity after therapy is a desirable goal as this results in improvement of PFS and OS.14, 15 Therefore, one of the key secondary objectives was to compare MRD negativity within each treatment group.
Patients and methods
Trial design
ADMIRE was a multicenter, randomized, controlled, open-label, parallel-group, phase IIB superiority trial assessing FCR (control) vs FCM-R (experimental) for previously untreated patients with CLL requiring treatment by IWCLL (International Workshop on Chronic Lymphocytic Leukemia) criteria.16 Patients were randomly allocated via a central computer-generated minimization programme that incorporated a random element 1:1 to receive oral fludarabine, cyclophosphamide and intravenous rituximab with or without intravenous mitoxantrone. Randomization was stratified to ensure balance for center, Binet stage (Progressive A or B, C), age group (⩽65, >65 years) and sex.
The primary objective of the trial was to assess whether the addition of mitoxantrone to FCR improved CR rates in patients with previously untreated CLL. The results would be used to determine whether a larger randomized phase III trial to formally assess survival was appropriate.
An independent Data Monitoring Committee (DMC) was established to review the safety and ethics of the trial. The DMC reviewed unblinded safety data on a 6-monthly basis and unblinded safety and trial progress reports on an annual basis. The DMC reported to an established trial steering committee (TSC) that provided general oversight for the trial.
The trial protocol was approved by the Leeds West Research Ethics Committee and the Medicines and Healthcare products Regulatory Agency. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The trial was registered as an International Standard Randomized Controlled Trial (ISRCTN42165735) and on the European Clinical Trials Database (EudraCT: 2008-006342-25).
Patients
The trial was planned to include 218 patients from hospitals around the UK. Eligible patients had: progressive CLL requiring treatment by IWCLL criteria;16 no prior treatment for CLL; World Health Organization (WHO) performance status (PS) 0–2; Binet stage progressive A, B or C; and provided written informed consent. Patients were not eligible if they had hepatitis B or C; an active secondary malignancy (excluding basal cell carcinoma of the skin); an active infection; or past history of anaphylaxis following exposure to rat- or mouse-derived complementarity determining region-grafted humanized monoclonal antibody. Patients with creatinine clearance >30 ml/min were allowed to enter the trial with guidance on dose reduction for fludarabine. Patients with a 17p deletion were eligible for enrollment due to lack of treatment options at the time of designing the trial. All patients provided written informed consent prior to trial enrollment and patients were able to withdraw from the trial at any time.
Treatment and assessments
Treatment with FCR or FCM-R was repeated every 28 days for a total of six cycles. Fludarabine and cyclophosphamide were administered orally at doses of 24 and 150 mg/m2/day, respectively, for the first 5 days of each cycle. These doses are pharmacologically equivalent to the doses used when FCR is given intravenously for CLL.17 This is in contrast to similar studies where intravenous doses of fludarabine and cyclophosphamide are used.1, 2, 3, 6 Mitoxantrone was administered intravenously on day 1 at a dose of 6 mg/m2 in the FCM-R group. Rituximab was administered intravenously at 375 mg/m2 on day 1 of cycle 1 and 500 mg/m2 in cycles 2–6. In participants with lymphocyte counts >25 × 109/l, the dose of rituximab was split to 100 mg on day 1 with the remaining dose given on day 2 to reduce the risk of infusion-related reactions. Participants unable to tolerate oral chemotherapy were permitted to receive equivalent intravenous doses of fludarabine (25 mg/m2/day for 3 days) and cyclophosphamide (250 mg/m2/day for 3 days). All participants were given allopurinol at least in cycle 1. Pneumocystis carinii pneumonia (PCP) and acyclovir prophylaxis and acyclovir were given throughout the treatment. Secondary prophylaxis with granulocyte colony-stimulating factor (G-CSF) was recommended for patients experiencing scheduled delays due to neutropenia. Appropriate dose reductions were recommended in patients with therapy-related cytopenias.
Participants were assessed for response at 3 months post-treatment and at 12, 18 and 24 months post-randomization in the absence of disease progression requiring treatment. Long-term annual follow-up for survival is being performed until death.
End points
The primary end point was CR rate (including CR with incomplete marrow recovery (CRi)) at 3 months post-treatment. Response was centrally assessed according to IWCLL criteria16 by two independent, experienced CLL hematologists blinded to treatment allocation. An independent arbiter reviewed discordant reports.
Secondary end points at 3 months post-treatment included: MRD negativity assessed in the bone marrow by highly sensitive multiparameter flow cytometry with a level of detection <1 CLL cell in 10 000 leukocytes;13 ORR defined as at least partial remission (PR); and safety and toxicity as graded by CTCAE (Common Terminology Criteria for Adverse Events) V3.0.18
Longer-term secondary end points included PFS, OS and time to MRD relapse in participants who became MRD negative.
Sample size
The sample size was based on testing the null hypothesis of no difference in CR rates between the treatment groups. The CR rate with FCR was estimated to be 50%, with a clinically important improvement considered to be 20%. With a two-sided 5% level of significance and 80% power, 103 participants were required in each group. Allowing for a 5% dropout rate, the recruitment target was 218 participants.
Statistical methods
All analyses were conducted on the intention-to-treat population, in which participants were included according to their randomized treatment. Safety analyses included participants according to treatment received. A two-sided 5% significance level was used for all formal efficacy end point comparisons.
Methods for handling missing end point data were prespecified and approved by the Chief Investigator. Participants with a missing assessment who died from CLL or treatment-related toxicity prior to their primary end point assessment or discontinued treatment early due to non-response or toxicity were treated as non-responders/MRD-positive. In the formal statistical analysis of the primary end point, for participants with at least a PR but missing trephine data to confirm a CR, imputation methods treated MRD-negative participants as having a CR and MRD-positive as not, although summaries also report the unimputed data. Participants without an available end point assessment were not included in the formal statistical analysis of the primary end point. This was appropriate as it can be assumed that data are missing completely at random, as assessments were most likely unavailable due to samples being unassessable or missed in error, rather than participant refusal due to level of response or treatment allocation. Sensitivity analyses assessed the robustness of the assumptions regarding missing primary end point data.
Multivariable binary logistic regression models compared CR rates, proportions with undetectable MRD (MRD negative) and ORR between the treatment groups, adjusted for the minimization factors, excluding center. Parameter estimates, s.e., odds ratios (OR) and corresponding P-values for the treatment effect are reported. The differences in proportions are reported with 95% confidence intervals (CIs) and corresponding P-values.
Kaplan–Meier curves are presented for the PFS and OS end points. Restricted mean survival time, used in the event of non-proportional hazards,19 estimated the area under the PFS curves, and treatment groups were compared using generalized linear regression, adjusted for the minimization factors, excluding center. Parameter estimates, s.e., ORs and corresponding P-values for the treatment effect are reported. Multivariable Cox regression analysis formally compared OS between treatment groups, hazard ratios (HRs), 95% CIs and corresponding P-values for the treatment effect are reported. Participants without evidence of an event at the time of analysis were censored at the last date they were known to be alive and event-free.
Safety analyses summarized the number of safety events occurring after randomization, including treatment-related mortalities (within 3 months post-treatment) and incidence of secondary cancers.
Prespecified exploratory subgroup analyses assessed the heterogeneity of the treatment effect among subgroups of interest for the primary end point, PFS and OS. Formal statistical testing between subgroups was not appropriate due to multiple testing errors and the reduced numbers in each subgroup. Subgroup analyses were interpreted with caution and treated as hypothesis generating.
Code availability
All statistical analyses were carried out using the SAS software 9.4 (SAS Institute Inc., Cary, NC, USA). Statistical analysis programs were validated but are not available publicly.
Results
Patient characteristics
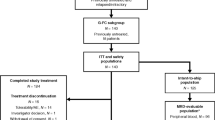
The CONSORT diagram20 (Figure 1) shows the flow of participants through the trial. A total of 420 patients were screened for eligibility. Of the 205 patients not randomized, the majority were clinically ineligible (n=112, 54.6%). Common reasons included: asymptomatic CLL, poor performance status, prior therapy for CLL, cardiac problems/unstable angina, second malignancy, or not having B-CLL.

CONSORT diagram.
In total, 215 participants were recruited between July 2009 and April 2012 (FCR: 107, FCM-R: 108) from 29 UK institutions with local ethical and management approval. The planned recruitment period ended before the target of 218 could be met. At the time of reporting, it has been approximately 7 years since the trial opened to recruitment, with a median follow-up of 5 years.
The baseline characteristics are displayed in Table 1. The median age was 62 years (range 33–77) with 74 participants (34.4%) aged >65 years. There was a male predominance (163 (75.8%)) and 27 participants (12.6%) were Binet stage progressive A, 111 (51.6%) stage B and 77 (35.8%) stage C. A majority of participants (124 (57.7%)) were WHO PS 0, with 83 (38.6%) PS 1 and 8 (3.7%) PS 2. Overall, 98 participants (45.6%) had B-symptoms (FCR: 51 (47.7%); FCM-R: 47 (43.5%)), while 123 (57.2%) had a β2-microglobulin concentration of ⩾4 mg/l and 30 (14.0%) had creatinine clearance levels of 30–60 mls/min. Of the evaluable participants, 14/203 (6.9%) had a 17p deletion (FCR: 9/100 (9.0%); FCM-R: 5/103 (4.9%)) and 38/203 (18.7%) an 11q deletion (FCR: 18/100 (18.0%); FCM-R: 20/103 (19.4%)). In all, 127/201 participants (63.2%) were considered to be ‘poorer risk’ in terms of VH mutational status, that is, VH unmutated or involving the VH3-21 gene (FCR: 68/101 (67.3%); FCM-R: 59/100 (59.0%)). Twenty participants (10.0%) presented with the VH3-21 gene (FCR: 14; FCM-R: 6).
Treatment
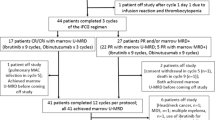
Of the 215 participants, 154 (71.6%) received 6 cycles of treatment (FCR: 82 (76.6%); FCM-R: 72 (66.7%)) (Table 2), and 24 (11.2%) received ⩽3 cycles of treatment (FCR: 11 (10.3%); FCM-R: 13 (12.0%)). Four participants did not receive any protocol treatment (FCR: 3 (2.8%); FCM-R: 1 (0.9%)), three did not meet the eligibility criteria and one participant allocated to receive FCR was removed by the treating clinician (Figure 1). Sixty-one participants (28.4%) discontinued treatment prematurely (FCR: 25 (23.4%); FCM-R: 36 (33.3%)) (Table 2). Reasons included: toxicity (n=43); progressive disease (n=2); stable disease with no/minimal response (n=2); ineligibility (n=4); participant choice (n=3); clinician decision (n=5); and other (n=2). Overall, 129 (60.0%) participants received G-CSF during treatment as recommended in the protocol as secondary prophylaxis, with a higher proportion in the FCM-R group (FCR: 60 (56.1%); FCM-R: 69 (63.9%)). Twenty participants unable to tolerate oral chemotherapy received equivalent intravenous doses (FCR: 8 (7.5%), FCM-R: 12 (11.1%)).
Efficacy
Overall, 8.4% (n=18) of participants were lost to follow-up for the primary end point (FCR: 10.3%; FCM-R: 6.5%), and reasons are presented in Figure 1. Of the 215 participants, 125 (58.1%) achieved a CR (FCR: 60 (56.1%); FCM-R: 65 (60.2%)) (Table 3). In the formal analysis of the primary end point, after imputation using MRD outcome, 137/197 (69.5%) achieved a CR, with a similar proportion in each treatment group (FCR: 67/96 (69.8%); FCM-R: 70/101 (69.3%)) (Table 3). The difference in response rates (FCM-R−FCR) was −0.5% (95% CI: −13.3, 12.4%), P-value=0.941. In the multivariable logistic regression analysis, the OR for achieving a CR with FCM-R compared with FCR was 0.97 (95% CI: 0.53, 1.79), P-value=0.932, concluding that the difference between the treatment groups is not significant at the 5% level. The sensitivity analyses (including comparison of CR rates prior to imputation) did not alter the findings.
There were no large differences in proportion of participants achieving a CR by gender (male: 100/148 (67.6%), female: 37/49 (75.5%)), age group (⩽65: 91/130 (70.0%), >65: 46/67 (68.7%)), Binet stage (Progressive A or B: 93/130 (71.5%), C: 44/67 (65.7%)) or creatinine clearance levels (ml/min) (30–60: 22/30 (73.3%), >60: 111/160 (69.4%)). A significantly higher proportion of participants who received >3 cycles of treatment achieved a CR (>3cycles: 135/183 (73.8%); ⩽3 cycles: 2/14 (14.3%); difference (95% CI): −59.5% (−78.9, −40.1%)). There were no large differences for the primary end point for those participants receiving G-CSF during treatment cycles 2–6 (G-CSF received: 81/121 (69.9%), no G-CSF: 51/71 (71.8%)).
Lower proportions of participants with a 17p deletion, 11q-deletion and ‘poorer risk’ VH mutational status achieved a CR (17pdel: 5/11 (45.5%); no 17pdel: 124/176 (70.5%)), (11qdel: 23/37 (62.2%); no 11qdel: 106/150 (70.7%)), (VH unmutated or VH3-21: 76/117 (65.0%); VH mutated: 52/69 (75.4%)).
Of the 215 participants, 191 (88.8%) achieved at least a PR (FCR: 93 (86.9%), FCM-R: 98 (90.7%)). Of the assessable participants, the ORR was 97.0% (191/197), with a similar proportion in each treatment group (FCR: 93/96 (96.9%), FCM-R: 98/101 (97.0%), with a difference (FCM-R−FCR) of 0.15% (95% CI: −4.6, 5.0%). A binary logistic regression analysis formally comparing the ORR between the treatment groups was unable to be performed owing to the small number of participants in the non-responders group.
Of the 215 participants, 101 (47.0%) achieved MRD negativity assessed in the bone marrow 3 month post-treatment (FCR: 54 (50.5%); FCM-R: 47 (43.5%) (Table 3). In the formal analysis of MRD (excluding participants with a missing MRD assessment), 101/184 (54.9%) achieved MRD negativity (FCR: 54/91 (59.3%), FCM-R: 47/93 (50.5%)). The difference in response rates (FCM-R−FCR) was −8.8% (95% CI: −23.1, 5.5%), P-value=0.230. In the multivariable logistic regression analysis, the adjusted OR for achieving MRD negativity with FCM-R compared with FCR was: 0.70 (95% CI: (0.39, 1.26), P=0.231), concluding that the difference between the treatment groups is not significant at the 5% level (Table 3).
At the time of analysis (4-year post-randomization of the final participant), 42 (19.5%) participants have died (FCR: 24 (22.4%), FCM-R: 18 (16.7%)), and 89 (41.4%) have either progressed or died (FCR: 44 (41.1%), FCM-R: 45 (41.7%)). Table 4 presents the primary cause of death by treatment group. Of the 42 participant deaths, 20 (47.6%) were due to CLL, that is, infection due to CLL, overwhelming tumor load or high-grade transformation of CLL (FCR: 13 (54.2%), FCM-R: 7 (38.9%)). Eight (19.0%) were treatment related, including treatment-related myelodysplastic syndrome/acute myeloid leukemia and infection due to treatment (FCR: 6 (25.0%), FCM-R: 2 (11.1%)). Figure 2 presents the PFS and OS Kaplan–Meier curves by treatment group. The mean PFS time up to a restricted time of 72 months postrandomization was 51.7 and 52.3 months in the FCR and FCM-R groups, respectively. The difference in the restricted mean survival between the treatment groups was not significant (FCM-R vs FCR: parameter estimate: 0.48, s.e.: 3.23, P=0.8823). For OS, the HR (FCM-R vs FCR) was not significant in the adjusted Cox regression model (HR and 95% CI: 0.75 (0.41, 1.39), P=0.3596).

Kaplan–Meier curves for PFS and OS. (a) PFS by treatment group. (b) OS by treatment group.
Of the 101 participants who were MRD negative in the bone marrow at 3 months post-treatment (Table 3), 23 (22.8%) have either relapsed at the MRD level in the peripheral blood or progressed (FCR: 11/54 (20.4%), FCM-R: 12/47 (25.5%)). The curves are not presented owing to the small number of events.
For the planned subgroup analyses, Kaplan–Meier curves demonstrated an improved PFS for participants who achieved a CR or MRD negativity at 3 months post-treatment and for those with a VH-mutated gene (and not VH3-21), that is, ‘standard risk’ patients (Figure 3). Both sensitivity analyses for CR status prior to imputation and subgroup analyses for OS show similar trends.

Kaplan–Meier curves for PFS subgroup analyses. (a) PFS by CR status at 3 months post-treatment. (b) PFS by MRD status at 3 months post-treatment (assessed in the bone marrow). (c) PFS by VH mutational risk status.
Safety and toxicity
The safety population included 212 participants (Figure 1). One hundred and fifty-six SAEs were reported from 97 (45.8%) participants, a lower proportion receiving FCR (41.9%) compared with FCM-R (49.5%). One hundred and sixteen serious adverse reactions (SARs) were reported from 76 (35.8%) participants (FCR: 55 events from 36 (34.3%); FCM-R: 61 events from 40 (37.4%)). The most commonly reported SARs, 65.5% of events (n=76), were infections and infestations. Ninety-two (43.4%) participants required hospitalization for a serious adverse event (SAE) (FCR: 43 (41.0%); FCM-R: 49 (45.8%)) (Table 5).
One suspected unexpected SAR, that is, a SUSAR, was reported was reported from a participant receiving all six cycles of FCM-R. They experienced prolonged myelosuppression and had a hypoplastic marrow on their 3-month post-treatment bone marrow aspirate. The event was suspected to be related to F, C and M.
Non-serious AEs were reported from 210 (99.1%) participants, with similar proportions in each treatment group. Of the 2914 AEs reported, 468 (16.1%) were graded as CTCAE grade ⩾3 (FCR: 222 (15.9%); FCM-R: 246 (16.2%); Table 5).
There was one treatment-related mortality reported within 3 months of the end of protocol treatment from a participant receiving FCR.
Within 5 years of participants ending treatment, 39 participants (18.4%) had been diagnosed with a secondary cancer (FCR: 19 (18.1%); FCM-R: 20 (18.7%)). The most commonly reported secondary cancers were non-melanoma skin cancers in 6.1% (n=13) of participants, followed by non-hematological solid tumors in 5.7% of participants (n=12) (Table 5). There have been two reports of myelodysplastic syndrome, one from each treatment group.
Discussion
This multicenter collaborative trial demonstrates that oral FCR results in extremely high response and MRD-negative rates (ORR: 97%, CR: 70%, MRD negativity: 59%). Trial follow-up is at a median of 5 years and there are a high number of censored observations, but to date, the PFS and OS are favorable compared with previous studies. The mean PFS for both trial arms is similar, with no significant difference. PFS was improved in participants achieving CR and MRD negativity. Participants with mutated VH genes (excluding VH3-21) had improved PFS compared with those with unmutated VH genes or using VH3-21. The PFS curves for VH-mutated genes (excluding VH3-21) plateau at a PFS probability of approximately 35%. Similar plateaus for PFS for this subgroup have been observed in previous studies.1, 21 The FCM-R group results appear equivalent, but the depth of responses was no higher with the addition of mitoxantrone to FCR (ORR: 97%; CR: 69%; MRD negativity: 51%). The median age of participants was 62 years, which is comparable to other front-line CLL trials of fludarabine-based therapies. In all, 89% of participants received greater than three cycles of treatment, and 72% of the participants received all six cycles of treatment. PCP and acyclovir prophylaxis was recommended for all participants. Secondary prophylaxis with G-CSF was administered to 60% of participants, enabling the delivery of a maximum number of treatment cycles. This may explain the high response and MRD-negative rates in our trial. The dose of fludarabine was reduced by 50% in participants with creatinine clearance between 30 and 60 mls/min. The 30 (14%) participants with creatinine clearance levels of 30–60 mls/min had a similar CR rate of 73.3% to those with levels >60 mls/min. This might suggest that selected participants considered unfit for FCR due to renal dysfunction can tolerate dose-modified FCR with high response rates.
The addition of mitoxantrone to FCR does not appear to have substantially increased toxicity rates, with 34.3% of participants experiencing a SAR with FCR compared with 37.4% with FCM-R. A similar proportion of grade 3 or 4 AEs were experienced in each treatment group (FCR: 15.9 vs FCM-R: 16.2%).
In summary, we have demonstrated that the addition of mitoxantrone to front-line FCR did not improve responses but slightly increased toxicity. In view of this, FCM-R will not be taken forward into a larger definitive phase III trial. The trial demonstrated that oral FCR given at an equivalent dose to intravenous FCR yields extremely high response rates compared with historical series and was well tolerated. This is consistent with the outcome of its companion trial ARCTIC comparing FCR with FCM-miniR (reported in the companion paper).22 The explanation for the high response rates is not certain but is possibly due to the fact that in the oral regime the same dose of chemotherapy is spread over 5 rather than 3 days and that the duration of therapy exposure per cycle may be critical. In addition, dose intensity was optimized by primary prophylaxis with acyclovir and co-trimoxazole and secondary prophylaxis with G-CSF. It was also possible to use dose-adjusted FCR for participants with impaired renal function.
FCR therefore remains the gold-standard therapy for CLL in participants considered fit for fludarabine-based therapy against which the novel targeted therapies must be tested, with oral administration of FC giving results at least as good as those obtained with intravenous administration.
References
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 2016; 127: 208–215.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010; 376: 1164–1174.
Tam CS, O'Brien S, Wierda W, Kantarjian H, Wen S, Do KA et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008; 112: 975–980.
Wilder DD, Ogden JL, Jain VK . Efficacy of fludarabine/mitoxantrone/dexamethasone alternating with CHOP in bulky follicular non-Hodgkin's lymphoma. Clin Lymphoma 2002; 2: 229–237.
Zinzani PL, Magagnoli M, Moretti L, Battista R, Ronconi F, De Renzo A et al. Fludarabine based chemotherapy in untreated mantle cell lymphomas: an encouraging experience in 29 patients. Haematologica 1999; 84: 1002–1006.
Bosch F, Ferrer A, Villamor N, Gonzalez M, Briones J, Gonzalez-Barca E et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: High response rate and disease eradication. Clin Cancer Res 2008; 14: 155–161.
Bosch F, Abrisqueta P, Villamor N, Terol MJ, Gonzalez-Barca E, Ferra C et al. Rituximab, fludarabine, cyclophosphamide, and mitoxantrone: a new, highly active chemoimmunotherapy regimen for chronic lymphocytic leukemia. J Clin Oncol 2009; 27: 4578–4584.
Bosch F, Ferrer A, Lopez-Guillermo A, Gine E, Bellosillo B, Villamor N et al. Fludarabine, cyclophosphamide and mitoxantrone in the treatment of resistant or relapsed chronic lymphocytic leukaemia. Br J Haematol 2002; 119: 976–984.
Hendry L, Bowen A, Matutes E, Swansbury J, Catovsky D . Fludarabine, cyclophosphamide and mitoxantrone in relapsed or refractory chronic lymphocytic leukemia and low grade non-Hodgkin's lymphoma. Leuk Lymphoma 2004; 45: 945–950.
Hillmen P, Cohen DR, Cocks K, Pettitt A, Sayala HA, Rawstron AC et al. A randomized phase II trial of fludarabine, cyclophosphamide and mitoxantrone (FCM) with or without rituximab in previously treated chronic lymphocytic leukaemia. Br J Haematol 2011; 152: 570–578.
Kwok M, Rawstron AC, Varghese A, Hillmen P . Minimal residual disease is a predictor for progression-free and overall survival in chronic lymphocytic leukemia (CLL) that is independent of the type or line of therapy. Blood 2009; 114: 226.
Rawstron AC, Kennedy B, Evans PAS, Davies FE, Richards SJ, Haynes AP et al. Quantitation of minimal disease levels in chronic lymphocytic leukemia using a sensitive flow cytometric assay improves the prediction of outcome and can be used to optimize therapy. Blood 2001; 98: 29–35.
Rawstron AC, Villamor N, Ritgen M, Boettcher S, Ghia P, Zehnder JL et al. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia 2007; 21: 956–964.
Böttcher S, Ritgen M, Fischer K, Stilgenbauer S, Busch RM, Fingerle-Rowson G et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol 2012; 30: 980–988.
Kwok M, Rawstron A, Varghese A, Evans P, O'Connor S, Doughty C et al. Independent prognostic significance of minimal residual disease status in chronic lymphocytic leukaemia. Lancet 383: S66.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111: 5446–5456.
Dearden CE, Richards S, Else M, Catovsky D, Hillmen P . A comparison of the efficacy and safety of oral and intravenous fludarabine in chronic lymphocytic leukemia in the LRF CLL4 trial. Cancer 2011; 117: 2452–2460.
Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events 2006. v3.0. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf.
Royston P, Parmar MKB . The use of restricted mean survival time to estimate the treatment effect in randomized clinical trials when the proportional hazards assumption is in doubt. Stat Med 2011; 30: 2409–2421.
Moher D, Schulz KF, Altman DG, Grp C . The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001; 357: 1191–1194.
Thompson PA, Tam CS, O'Brien SM, Wierda WG, Stingo F, Plunkett W et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 2016; 127: 303–309.
Howard DR, Munir T, McParland L, Rawstron AC, Milligan D, Schuh A et al. Results of the randomized phase IIB ARCTIC trial of low dose rituximab in previously untreated CLL. Leukaemia 2017; e-pub ahead of print 24 March 2017; doi:10.1038/leu.2017.96.
Acknowledgements
ADMIRE is an National Institute for Health Research (NIHR) Portfolio Study developed in association with the NCRI CLL Subgroup and funded by Roche Products Limited. The views and opinions expressed there in are those of the authors and do not necessarily reflect those of Roche, NIHR or the NHS. The authors thank all patients and hospital staff who contributed to this study. In addition, they acknowledge the invaluable support provided by the independent DMC and TSC and Roche Products Limited for providing rituximab for the trial as well as an unrestricted grant to support the running of the trial.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Professor Hillmen received research funding and speakers’ fees from Roche Products Limited. Dr Rawstron, Dr Munir, Dr Fegan and Dr Hamblin reports personal fees from Roche Products Limited. Dr Bloor has also reported personal, consultancy/advisory and speaker’s fees from Roche Products Limited. Professor Gribben reports personal fees and expenses from Roche Products Limited. The other authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Munir, T., Howard, D., McParland, L. et al. Results of the randomized phase IIB ADMIRE trial of FCR with or without mitoxantrone in previously untreated CLL. Leukemia 31, 2085–2093 (2017). https://doi.org/10.1038/leu.2017.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.65
This article is cited by
-
Combined analysis of IGHV mutations, telomere length and CD49d identifies long-term progression-free survivors in TP53 wild-type CLL treated with FCR-based therapies
Leukemia (2022)
-
Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features
Nature Genetics (2022)
-
Measurable residual disease in chronic lymphocytic leukemia: expert review and consensus recommendations
Leukemia (2021)
-
Telomere length predicts for outcome to FCR chemotherapy in CLL
Leukemia (2019)
-
Results of the randomized phase IIB ARCTIC trial of low-dose rituximab in previously untreated CLL
Leukemia (2017)
