
Abstract
Tenascin-C is expressed in choroidal neovascular (CNV) membranes in eyes with age-related macular degeneration (AMD). However, its role in the pathogenesis of CNV remains to be elucidated. Here we investigated the role of tenascin-C in CNV formation. In immunofluorescence analyses, tenascin-C co-stained with α-SMA, pan-cytokeratin, CD31, CD34, and integrin αV in the CNV membranes of patients with AMD and a mouse model of laser-induced CNV. A marked increase in the expression of tenascin-C mRNA and protein was observed 3 days after laser photocoagulation in the mouse CNV model. Tenascin-C was also shown to promote proliferation and inhibit adhesion of human retinal pigment epithelial (hRPE) cells in vitro. Moreover, tenascin-C promoted proliferation, adhesion, migration, and tube formation in human microvascular endothelial cells (HMVECs); these functions were, however, blocked by cilengitide, an integrin αV inhibitor. Exposure to TGF-β2 increased tenascin-C expression in hRPE cells. Conditioned media harvested from TGF-β2-treated hRPE cell cultures enhanced HMVEC proliferation and tube formation, which were inhibited by pretreatment with tenascin-C siRNA. The CNV volume was significantly reduced in tenascin-C knockout mice and tenascin-C siRNA-injected mice. These findings suggest that tenascin-C is secreted by transdifferentiated RPE cells and promotes the development of CNV via integrin αV in a paracrine manner. Therefore, tenascin-C could be a potential therapeutic target for the inhibition of CNV development associated with AMD.
Similar content being viewed by others


Main
Age-related macular degeneration (AMD) is one of the leading causes of vision reduction in older people in developed countries.1 The decrease in visual acuity is usually attributed to choroidal neovascularization (CNV), a pathological invasion of new blood vessels into the subretinal space from the choriocapillaris through the Bruch’s membrane. The development of these vessels is accompanied by retinal edema, retinal detachment, and hemorrhage, which destroys photoreceptor cells.2
Recently, a number of studies have focused on the effects of anti-vascular endothelial growth factor (VEGF) treatment on CNV in eyes with AMD.3, 4, 5, 6 Monthly intravitreal injections of ranibizumab, an anti-VEGF agent, prevented vision reduction in nearly 95% of the AMD patients, and significantly improved the vision in 40% of the patients, for 2 years.3 However, the use of anti-VEGF drugs to maintain vision over long periods of time causes problems in tolerance or tachyphylaxis, which is a reduction in drug efficacy following repeated administration.7, 8, 9 Approximately 18.3% of all AMD patients treated with anti-VEGF drugs reportedly develop geographic atrophy within 2 years of beginning the treatment.10 Therefore, many research efforts are now directed towards the identification of new agents that could be effective in the treatment of CNV.
We and others have reported that tenascin-C promotes the retinal neovascularization associated with proliferative diabetic retinopathy.11, 12, 13 Tenascin-C is a large hexameric extracellular glycoprotein composed of 180–320 kDa monomers. It consists of four major domains: a globular N-terminal assembly domain, a 14.5 epidermal growth factor-like repeats domain, a fibronectin type III-like repeat domain, and a fibrinogen-like sequence. Fibronectin type III-like repeats of tenascin-C affect cell adhesion via integrin receptors and Annexin II.14 Tenascin-C is chiefly expressed during development, but is also present at low levels in most adult tissues. However, tenascin-C is continuously upregulated during active inflammation and tissue repair.15 Several studies have reported that tenascin-C plays a vital role in cardiac ischemia,16, 17, 18 tumor angiogenesis, and metastasis.19, 20, 21, 22 Moreover, it has been reported that CNV membranes obtained from patients with AMD containing retinal pigment epithelial (RPE) cells express tenascin-C.23, 24
We have recently reported that periostin, a matricellular protein produced by RPE cells, promotes CNV formation.25 However, the angiogenic processes mediated by tenascin-C in the pathogenesis of CNV have not been elucidated.
Therefore, the purpose of this study was to investigate the expression and function of tenascin-C in human CNV membranes in an in vivo mouse CNV model as well as RPE and vascular endothelial cells in vitro. We also evaluated the efficacy of tenascin-C as a potential therapeutic target using acute tenascin-C blockade in a mouse CNV model.
Materials and methods
Human Specimens
This study was approved by the Ethics Committee of Kyushu University Hospital; the surgical specimens were handled in accordance with the Declaration of Helsinki. Signed informed consent forms were obtained from all patients prior to the surgical procedure. CNV membranes were surgically removed from three eyes of three patients with wet-type AMD. The ages of these patients at the time of pars plana vitrectomy ranged from 62 to 81 years.
Animals
All animal experiments were approved by the Ethics Committee for Animal Experiments of the Kyushu University Graduate School of Medical Sciences. C57BL/6J mice were purchased from CLEA Japan (Tokyo, Japan) and tenascin-C knockout mice were purchased from the RIKEN BioResource Center (Ibaraki, Japan). Tenascin-C knockout mice are also known as rd8/rd8 mice as they carry the Crb1 gene, which is associated with retinal degeneration;26 therefore, the rd8/rd8 mutation was bred out by backcrossing the knockout mice with the C57BL/6J line.
Mouse Model of CNV
The Bruch’s membrane was ruptured by laser photocoagulation to generate CNV in mice.27 Briefly, 7-week-old male C57BL/6J and tenascin-C knockout mice were anesthetized, and their pupils were dilated with 0.5% tropicamide. Four burns were produced on each retina by a 532 nm diode laser (75 μm spot size, 0.1 s duration, 100 mW) of a slit-lamp-delivery system in a photocoagulator (Novus Verdi; Coherent Inc., Santa Clara, CA, USA).
The effect of tenascin-C siRNA was evaluated in vivo by injecting tenascin-C or control siRNA (#4390843: Thermo Fisher Scientific, Waltham, MA, USA), with randomly scrambled sequences into the eye immediately after laser photocoagulation.
Immunofluorescence Staining
Immunohistochemistry was performed as described previously.28, 29 Briefly, as the number of AMD patients for surgery was limited, CNV membranes from clinical samples were embedded in paraffin and cut into 3-μm sections for long-term sample preservation. The sections were deparaffinized, rehydrated, blocked, and finally incubated overnight with primary antibodies. The sections were subsequently incubated with secondary antibodies for 30 min at room temperature. For immunofluorescence analysis in mice, the eyeballs of mice with photocoagulation (5 days after laser-treatment) were embedded in OCT compound (Tissue-Tek; Sakura Seiki, Tokyo, Japan) and frozen on dry ice. The frozen mouse eyes were cryosectioned to a thickness of 10 μm, fixed in −20 °C acetone for 10 min, blocked with 5% skim milk, and subsequently incubated overnight with the primary antibody. Finally, the sections were incubated with the secondary antibody for 20 min at room temperature. The antibodies are listed in the Supplementary Tables 1 and 2. The nuclei were counterstained with Hoechst 33342 (H3570: 1:400 dilution; Life Technologies, Gaithersburg, MD, USA), and the sections were examined with a fluorescence microscope (BZ-9000; Keyence, Osaka, Japan).
Quantification of CNV Volume in Mouse Model
The eyes were removed 7 days after photocoagulation and fixed in 4% paraformaldehyde for 1 h. The cornea, lens, and retina were removed, and the choroid/sclera segment was fixed for 24 h in 4% paraformaldehyde. The tissue was exposed to methanol for 20 min, treated with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) in PBS for 1 h, incubated in fluorescein-labeled isolectin B4 (FL1201; 1:200 dilution; Vector Laboratories, Burlingame, CA, USA) at room temperature for 1.5 h, and then flat-mounted on glass slides. Choroidal flat mounts were observed with a laser scanning confocal microscope (Nikon A1R; Nikon, Tokyo, Japan), and the volume at each burn site was measured using the NIS-Elements software (Nikon).
Cell Culture
Human retinal pigment epithelial cells (hRPE, 00194987; Lonza Walkersville, Walkersville, MD, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS). Human dermal microvascular endothelial cells (HMVECs, CC-2543; Lonza Walkersville) were cultured in EGM-2MV BulletKit (CC-3202; Lonza Walkersville).
The hRPE cells were seeded in collagen-coated 24-well plates for mRNA assays and 6-well plates for protein assays. The cells were starved in serum-free DMEM for 24 h, and subsequently treated with or without TGF-β2 (SRP3170; Sigma-Aldrich) for 24 h. mRNA was then extracted from these cells using TRIzol reagent (Life Technologies). The supernatant (containing the proteins) was collected after a 48-h incubation period, and total cell lysate was extracted with a lysis buffer.
Cilengitide (10 μM; Selleckchem, Houston, TX, USA) was added at the time of stimulation in all subsequent assays to inhibit integrin αVβ3 and αVβ5.
Real-Time qRT-PCR
The tenascin-C (and GAPDH control) mRNA expression was quantified by qRT-PCR, as described previously.25, 30 Briefly, total RNA was extracted and reverse-transcribed with a first-strand cDNA synthesis kit (Roche, Mannheim, Germany). The level of gene expression was determined by real-time RT-PCR, using a Roche LightCycler 96 (Roche). The quantification was carried out using either a TaqMan-probe or SYBR Green. The primers and annealing temperatures used are listed in Supplementary Table 3.
Enzyme-Linked Immunosorbent Assay (ELISA)
The protein concentration of tenascin-C was measured by sandwich ELISA, using a standard ELISA kit (27767; IBL, Gunma, Japan) in compliance with the manufacturer protocol. The samples were added to a buffer solution in 96-well plates and incubated for 1 h at room temperature. The solution was aspirated and washed. The wells were then incubated with horseradish peroxidase-conjugated mouse monoclonal antibody against tenascin-C (4F10TT) for 30 min at 4 °C. The solution was aspirated and washed again. The substrate solution was added to each well and the plate was incubated for 30 min at room temperature. The stop solution was added to each well and the plate was ready with a microplate reader (ImmunoMini NJ-2300; NJ InterMed, Tokyo, Japan) at 450 nm.
Western Blot Analysis
The protein samples were separated on a NuPAGE 3–8% Tris-Acetate gel (EA0375; Life Technologies) and the blots were incubated with a monoclonal antibody against tenascin-C (4F10TT: 10337: 1:3000; IBL). The blot was developed by an immunoperoxidase method (Envision+; Dako, Glostrup, Denmark) and subsequently visualized using a SuperSignal West Femto maximum sensitivity substrate (#34095; Thermo Fisher Scientific). Lane-loading differences were normalized by blotting the membranes with an antibody against β-actin (#4970: 1:5000; Cell Signaling Technologies, Beverley, MA, USA).
Cell Proliferation Assay
Proliferation of the hRPE cells and HMVECs was analyzed by bromodeoxyuridine (BrdU)-incorporated ELISA (Roche) according to the manufacturer’s protocol, with a 1.5-h BrdU incubation.
Cell Adhesion Assay
The cell adhesion assay was performed as described in a previous report.31 Briefly, hRPE cells were trypsinized and resuspended in serum-free DMEM containing recombinant tenascin-C. Cell suspension (1 × 104 cells/200 μl) was added to each fibronectin-coated well (F2006: 10 μg/ml; Sigma-Aldrich) and allowed to attach for 1 h. HMVECs (1 × 104 cells/200 μl) were resuspended in EBM-2 media with 0.5% FBS, containing recombinant tenascin-C, and allowed to attach for 12 h. The cells were gently washed twice with PBS, fixed with 4% PFA, and stained with Hoechst 33342 (H3570, 1:1000 dilution; Life Technologies) for 15 min. The stained cells were counted with a 4 × objective lens in four fields, and the data were analyzed with the ImageJ software (National Institute of Health, Bethesda, MD, USA).
Cell Migration Assay
The effect of tenascin-C on the migration of HMVECs was examined by performing transwell assays using cell culture inserts with a pore size of 8 μm (Corning, Corning, NY, USA). HMVECs were suspended at a concentration of 5 × 104 cells/ml in EBM-2 media, supplemented with 0.5% FBS and recombinant tenascin-C. To assess cell migration, HMVECs stimulated with tenascin-C were placed in the upper chamber and allowed to migrate to the reverse side of the membrane. The inserts were fixed with 4% PFA and stained with Hoechst 33342 (H3570, 1:1000 dilution; Life Technologies) for 15 min. The stained cells were counted in four random fields under a 20 × objective lens, and the data were analyzed using ImageJ software (National Institute of Health).
Tube Formation Assay
HMVECs were plated at a concentration of 1 × 104 cells/well in 96-well plates pre-coated with Matrigel Basement Membrane Matrix (BD Bioscience), in EBM-2 media containing 0.5% FBS and recombinant tenascin-C. The cells were subsequently incubated for 12 h at 37 °C. The data were analyzed using ImageJ software (National Institute of Health).
Preparation of hRPE-Conditioned Medium
hRPE-conditioned medium was produced by culturing hRPE cells in 6-well plates to a confluence of 70–80%. Tenascin-C siRNA (10 nM) or control siRNA (10 nM) was mixed with 4 μl lipofectamine RNAiMAX (Thermo Fisher Scientific) in 2 ml Opti-MEM (Thermo Fisher Scientific). This mixture was added to hRPE cells and the cells were incubated for 24 h. Recombinant TGF-β2 was added at a concentration of 10 ng/ml per well and incubated for 24 h. The composite transfection medium was removed and replaced with serum-free EBM-2. After 24 h, conditioned medium was collected from the 6-well plates and filtered through 40-μm filters (Corning).
Statistical Analyses
All results are expressed as the means±standard errors of means. The differences among groups were analyzed by two-tailed Student’s t tests or one-way analysis of variance (ANOVA) with Dunnett’s tests or Tukey tests. Statistical analyses were performed with a commercial statistical software package (JMP v.11.0; SAS Institute, Cary, NC, USA). A P-value <0.05 was taken to be statistically significant.
Results
Tenascin-C is Expressed by RPE Cells Transdifferentiated Into Myofibroblasts in CNV Membranes
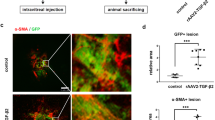
The location of tenascin-C in CNV membranes obtained from the eyes of AMD patients were determined by staining the sections with antibodies against tenascin-C, α-smooth muscle actin (α-SMA), pan-cytokeratin, and CD34, which are markers for myofibroblasts, RPE cells, and vascular endothelial cells, respectively. In the CNV membranes, RPE cells and myofibroblasts were colocalized with tenascin-C. Alternately, vascular endothelial cells were localized around tenascin-C (Figure 1a).

(a) Representative photographs of triple immunofluorescence staining for tenascin-C, α-SMA, and CD34 in a CNV membrane excised from an AMD patient. Tenascin-C staining was observed in α-SMA- and pan-cytokeratin-positive cells, and around CD34 positive cells. (b) Triple immunofluorescence staining for tenascin-C, pan-cytokeratin, α-SMA, CD31, F4/80, and integrin αV in lesions observed in a wild-type laser-induced CNV model 5 days post-laser treatment. Pan-cytokeratin-positive cells were stained with α-SMA. Tenascin-C co-stains with pan-cytokeratin, α-SMA, CD31, and integrin αV, but not with F4/80. Nuclei are stained blue. Scale bar, 50 μm.
Tenascin-C is Expressed by RPE Cells Transdifferentiated Into Myofibroblasts in a Laser CNV Mouse Model
The location of tenascin-C in CNV lesions was further determined by immunofluorescent staining for tenascin-C, pan-cytokeratin, α-SMA, CD31, F4/80, and integrin αV in the mouse CNV model 5 days after the retinal laser photocoagulation. CD31, F4/80, and integrin αV are markers for vascular endothelial cells, macrophages, and the tenascin-C receptor, respectively. The RPE cells were found to colocalize with tenascin-C as well as α-SMA, whereas vascular endothelial cells partly colocalized with tenascin-C and integrin αV. In contrast, macrophages colocalized weakly with tenascin-C. These findings indicate that tenascin-C is predominantly expressed in myofibroblasts transdifferentiated from RPE cells, and is bound to vascular endothelial cells via integrin αV in the CNV lesions (Figure 1b)
Synthesis and Secretion of Tenascin-C is Induced in Mouse CNV Model
The time course of tenascin-C expression during CNV formation was confirmed by evaluating the mRNA and protein expression of tenascin-C in RPE-choroid tissues obtained from the mouse CNV model at different times. Real-time qRT-PCR showed that the expression of tenascin-C was highly upregulated in the RPE-choroid at days 1 and 3 after laser coagulation, and decreased thereafter (Figure 2a). The kinetics of protein expression were similar, with peak levels observed at day 3, and markedly reduced protein levels from day 5 (Figures 2b and c). The 4F10TT antibody, which recognizes all tenascin-C variants, identified two isoforms of tenascin-C (210 and 250 kDa). Both the larger and smaller tenascin-C isoforms were expressed in the mouse CNV model.

Kinetics of changes in mRNA (a) and protein (b) levels of tenascin-C in the RPE-choroid complex obtained from a mouse CNV model (mean±s.e.m.; n=4 eyes per group). (a and b) Tenascin-C expression is significantly increased and peaked 3 days after laser photocoagulation. (c) Western blots of tenascin-C in the RPE-choroid complex of a mouse CNV model. The mouse monoclonal anti-tenascin-C clone 4F10TT antibody displayed 210 and 250 kDa isoforms of tenascin-C, and a similar trend. (d) Expression of tenascin-C mRNA normalized to GAPDH in hRPE cells stimulated with the indicated concentrations of transforming growth factor-β2 (TGF-β2) (mean±s.e.m.; n=4 per group). (e) ELISA of tenascin-C in the cell lysates and supernatants of hRPE cells stimulated with the indicated concentrations TGF-β2 (mean±s.e.m.; n=4 per group). (d, e) TGF-β2 significantly induces the expression of tenascin-C mRNA and protein, in a dose-dependent manner. (f) Western blot analysis of tenascin-C and β-actin in hRPE cells stimulated by the indicated concentrations of TGF-β2. The mouse monoclonal anti-tenascin-C clone 4F10TT antibody showed two isoforms of tenascin-C (210 and 300 kDa) and a similar trend. β-actin was used as the loading control. *P<0.05, **P<0.01, ANOVA/Dunnett’s test, compared to the control.
Synthesis and Secretion of Tenascin-C Is Also Induced in Transdifferentiated hRPE Cells Stimulated by TGF-β2
Based on the expression of tenascin-C in α-SMA-positive RPE cells, we hypothesized that TGF-β2-induced transdifferentiation of RPE cells into myofibroblasts might lead to tenascin-C production. This theory was tested by evaluating the expression of tenascin-C in hRPE cells exposed to recombinant TGF-β2. We have shown that TGF-β2 caused the cells to become elongated, spindle-shaped, and flat, resembling fibroblasts.32 Real-time qRT-PCR analysis showed that 1, 3, and 10 ng/ml TGF-β2 induced an upregulation in the mRNA and protein expression of tenascin-C in hRPE cells in a dose-dependent manner (Figures 2d and e). Consistently, western blot analysis indicated that tenascin-C protein expression was upregulated in the cell lysates of hRPE cells exposed to TGF-β2 (Figure 2f). The 4F10TT antibody identified two isoforms of tenascin-C, weighing 210 and 300 kDa. Both of these isoforms were expressed in hRPE cells.
Tenascin-C Promotes Proliferation and Reduces Adhesion of hRPE Cells
The effect of tenascin-C on hRPE cell function was tested by evaluating the effect of tenascin-C on hRPE cell proliferation and adhesion. Incubation with 30, 100, and 300 μg/ml tenascin-C significantly promoted hRPE cell proliferation, as indicated by increased BrdU incorporation, in a dose-dependent manner (Figure 3a). Recombinant tenascin-C also significantly inhibited the adhesion of hRPE cells to fibronectin-coated wells, which was indicated by the number of stained cells (Figures 3b and c). These results demonstrated that tenascin-C promotes proliferation and reduces adhesion of hRPE cells in an autocrine manner.

Effect of tenascin-C on proliferation (a) and adhesion (b and c) of hRPE cells and proliferation (d), adhesion (e and f), migration (g), and tube formation (h and i) of HMVECs. (a) hRPE cell proliferation was assessed by BrdU incorporation (mean±s.e.m.; n=4 per group). hRPE cell proliferation was significantly increased by tenascin-C in a dose-dependent manner. (b and c) hRPE cells were incubated with the indicated concentrations of tenascin-C or TGF-β2 (positive control) in fibronectin-coated 96-well plates for 1 h (mean±s.e.m.; n=4 per group) to analyze the cell adhesion. hRPE cell adhesion was significantly upregulated by TGF-β2 (10 ng/ml) and downregulated by tenascin-C (300 ng/ml). Photographs were taken with a 20 × objective after Hoechst 33342 staining. (d) HMVECs were incubated with the indicated concentrations of tenascin-C for 24 h to assess proliferation (mean±s.e.m.; n=4 per group). (e and f) HMVECs were incubated with the indicated concentrations of tenascin-C in fibronectin-coated 96-well plates for 12 h to assess adhesion (mean±s.e.m.; n=4 per group). (g) HMVECs stimulated with tenascin-C were placed in the upper chamber and allowed to migrate to the reverse side of the membrane to assess the cell migration (mean±s.e.m.; n=4 per group). (h and i) HMVECs were seeded on a basement membrane (BM) matrix with the indicated concentrations of tenascin-C (mean±s.e.m.; n=6 per group). Tubes formed by HMVECs were photographed after incubating for 12 h (4 × objective). (d–i) Tenascin-C significantly increased the HMVEC proliferation, adhesion, migration, and tube formation, in a dose-dependent manner. Values are expressed as a percentage relative to the untreated control. *P<0.05, **P<0.01, ANOVA/Dunnett’s test.
Tenascin-C Promotes HMVEC Proliferation, Adhesion, Migration, and Tube Formation
The contribution of tenascin-C to angiogenesis of HMVECs, including cell proliferation, adhesion, migration, and tube formation, was also tested. Exposure to 100 and 300 ng/ml tenascin-C significantly upregulated the HMVEC proliferation (Figure 3d); additionally, 30, 100, and 300 ng/ml recombinant tenascin-C significantly enhanced the adhesion of HMVECs to fibronectin-coated wells in a dose-dependent manner (Figures 3e and f). In the cell migration assay, 300 ng/ml tenascin-C was shown to significantly promote cell migration (Figure 3g), whereas the same quantity of tenascin-C was shown to significantly promote tube formation in the tube formation assay (Figures 3h and i).
Tenascin-C-Dependent Angiogenesis is Mediated by Integrin αV
To determine whether the tenascin-C-dependent HMVEC proliferation, adhesion, migration, and tube formation are mediated by αV integrin, we evaluated the inhibitory effect of cilengitide, an integrin αVβ3 and integrin αVβ5 inhibitor, on HMVECs. Integrin αV inhibition almost completely suppressed the tenascin-C-induced proliferation, adhesion, migration, and tube formation in HMVECs (Figures 4a–f). These results indicated that integrin αV mediates the tenascin-C stimulation of proliferation, adhesion, migration, and tube formation in HMVECs.

Inhibitory effect of cilengitide on tenascin-C-induced proliferation, adhesion, migration, and tube formation in HMVECs. (a) HMVECs were incubated with tenascin-C (300 ng/ml) and cilengitide (10 μM) for 24 h. (b and c) Cell adhesion was assessed by incubating the HMVECs with tenascin-C (300 ng/ml) and cilengitide (10 μM) on fibronectin-coated 96-well plates for 12 h (mean±s.e.m.; n=4 per group). (d) The cell migration was assessed placing HMVECs stimulated with tenascin-C (300 ng/ml) and cilengitide (10 μM) in the upper chamber and allowing the cells to migrate to the other side of the membrane (n=4 per group). (e and f) HMVECs were seeded on BM matrix with tenascin-C (300 ng/ml) and cilengitide (10 μM) (mean±s.e.m.; n=6 per group). Tubes formed by HMVECs were photographed after a 12-h incubation period (4 × objective). (a–f) Cilengitide significantly inhibited tenascin-C-induced proliferation, adhesion, migration, and tube formation of HMVECs. Values are expressed as a percentage relative to the control. *P<0.05, **P<0.01, ANOVA/Dunnett’s test.
Tenascin-C Secreted from Transdifferentiated hRPE Cells Promotes Angiogenesis
We hypothesized that tenascin-C secreted from transdifferentiated hRPE cells induces angiogenesis in a paracrine manner. This hypothesis was tested using a synthesized tenascin-C siRNA (Supplementary Table 4). To confirm the inhibitory effect of tenascin-C siRNA on the expression of tenascin-C in hRPE cells, we examined the production of tenascin-C in hRPE cells cultured with tenascin-C siRNA or control siRNA in the presence or absence of TGF-β2, which is expressed in CNV membranes.33 Without TGF-β2 stimulation, we observed no significant difference between cells transfected with tenascin-C or control siRNA. In contrast, in the presence of TGF-β2 stimulation, tenascin-C siRNA significantly inhibited the expression of tenascin-C (Figure 5a).

Inhibitory effect of tenascin-C siRNA on HMVEC proliferation and tube formation, induced by conditioned medium of hRPE cells treated with TGF-β2. (a) Tenascin-C expression in the conditioned medium of hRPE cells transfected with tenascin-C siRNA. Following transfection with tenascin-C or control siRNA (10 nM), hRPE cells were treated with or without TGF-β2 (10 ng/ml) for 24 h (mean±s.e.m.; n=4 per group). Tenascin-C siRNA significantly reduced the concentration of tenascin-C in the conditioned medium of hRPE cells treated with TGF-β2. (b) HMVECs were incubated to assess proliferation in hRPE-conditioned medium for 24 h. (c and d) HMVECs were seeded on BM matrix in hRPE-conditioned medium (mean±s.e.m.; n=6 per group). Tube formed by HMVECs were photographed after a 12-h incubation period (4 × objective). (b–d) HMVECs cultured in hRPE-conditioned medium transfected with tenascin-C siRNA after TGF-β2 stimulation showed a significant reduction in proliferation and tube formation compared with the control cells. Values are expressed as a percentage relative to the control. *P<0.05, **P<0.01, ANOVA/Tukey test.
To confirm that tenascin-C secreted from hRPE cells can promote CNV, we assessed the proliferation and tube formation of HMVECs cultured in hRPE-conditioned medium. hRPE-conditioned medium pretreated with TGF-β2 significantly promoted HMVEC proliferation and tube formation on Matrigel (Figures 5b–d). In contrast, HMVECs cultured in hRPE-conditioned medium pretreated with tenascin-C siRNA and TGF-β2 showed a significant reduction in proliferation and tube formation, compared with those cultured in hRPE pretreated with control siRNA and TGF-β2 (Figures 5b–d). These results indicated that tenascin-C from transdifferentiated hRPE cells facilitates CNV formation.
Disruption of Tenascin-C Suppresses CNV Formation in a Mouse CNV Model
The involvement of tenascin-C in CNV formation was confirmed by measuring the volume of CNV in the laser-induced tenascin-C knockout and wild-type C57BL/6J CNV mouse models. The CNV volume, measured in lectin-stained choroidal flat mounts, was significantly reduced to 53% in tenascin-C knockout mice compared with that seen in wild-type mice at day 7 (Figures 6a and b).

Volume of CNV formed after laser photocoagulation in tenascin-C knockout mice and mice treated with tenascin-C siRNA. (a) Analysis of the volume of CNV lesions 7 days after photocoagulation in wild-type and tenascin-C knockout mice (mean±s.e.m.; n=8 eyes per group). (b) Representative projections (green) and 3D images (red) of CNV lesions. Scale bar, 100 μm. (a and b) CNV volume was significantly smaller in the eyes of tenascin-C knockout mice compared with that in wild-type mice. (c) Analysis of the volume of CNV lesions 7 days after vitreous injection of control siRNA or tenascin-C siRNA (mean±s.e.m.; n=8 eyes per group). (d) Representative projections (green) and 3D images (red) of CNV lesions. Scale bar, 100 μm. (c and d) CNV volume was significantly lower in eyes treated with tenascin-C siRNA (10 and 100 nM) compared with that in eyes treated with control siRNA. *P<0.05, **P<0.01, Student’s t test or ANOVA/Dunnett’s test.
Finally, the possible therapeutic effect of tenascin-C siRNA was assessed by administering C57BL/6J mice with an intravitreous injection of 1, 10, and 100 μM tenascin-C or control siRNA following the laser injury. Ten and hundred μM tenascin-C siRNA induced a significant reduction in CNV volume (63 and 51%) compared with that induced by the control siRNA (Figures 6c and d).
Discussion
Our results showed that the induction of tenascin-C in the laser-induced mouse CNV model reached a peak at day 3 post-laser photocoagulation. Tenascin-C is expressed by RPE cells that have transdifferentiated into myofibroblasts in both the laser-induced mouse CNV model and CNV membranes from AMD patients. Together with the proliferation, migration, and tube formation of HMVECs, tenascin-C may be chronically expressed and may promote CNV membrane formation in AMD patients.
Several studies have reported that myofibroblasts are the major source of tenascin-C in the heart, colon, and liver.34, 35, 36 Consistent with this data, the immunohistochemical experiments performed in this study showed that tenascin-C was colocalized in the myofibroblasts and RPE cells of CNV membranes. Previous immunohistochemical findings in eyes with AMD showed intracellular distribution of TGF-β2 in RPE cells, the predominant isoform in the retina, RPE cells, and CNV membranes.33, 37 In addition, TGF-β2 is a cytokine of M2 macrophages identified in the CNV membrane.38, 39 As TGF-β2 contributes to the transdifferentiation of RPE cells into myofibroblasts,40, 41 TGF-β2 may be a potent inducer of tenascin-C in CNV membranes. Our in vitro assays showed that TGF-β2 induced the expression of tenascin-C in hRPE cells, which in turn promoted angiogenesis. These results demonstrate that myofibroblasts transdifferentiated from RPE cells via RPE- and/or M2 macrophage-derived TGF-β2 and continuously produced tenascin-C, resulting in CNV formation in a paracrine manner.
We also found that tenascin-C inhibited the adhesion of hRPE cells and promoted the proliferation of hRPE cells and vascular endothelial cells. Together with the proliferation, migration, and tube formation of HMVECs, these results suggest that tenascin-C promotes tissue remodeling close to the RPE-choroid interface by weakening the RPE cell adhesion to Bruch’s membrane, and facilitates the invasion of vessels, to form CNV in the subretinal space. Our findings are consistent with a previously proposed hypothesis that the cell and cytokine activity during CNV formation is very similar to that of wound healing.42 Active phase CNV contains a number of RPE cells, fibroblasts, and macrophages, which produce proangiogenic cytokines and extracellular matrix proteins, including tenascin-C and periostin.25, 42
In the immunohistochemical studies, vascular endothelial cells were co-stained with tenascin-C and integrin αV. Integrin αV is expressed in many types of cells. Integrin αVβ3 is a well-known receptor that is specifically involved in CNV43 and is one of the tenascin-C-binding receptors on vascular endothelial cells.11 Our results demonstrated that cilengitide inhibited cellular proliferation, adhesion, migration, and tube formation in HMVECs treated with tenascin-C. This was consistent with the results of a previous study where tenascin-C was shown to bind to human umbilical vein endothelial cells in the presence of integrin αVβ3.44 Together, these findings suggest that tenascin-C promotes CNV formation by binding to integrin αVβ3.
There were some limitations to our study. Although laser-induced mouse models are widely used as models of CNV, it is an acute injury model and therefore may not be an accurate model of CNV in AMD because the latter CNV occurs as a secondary response to chronic inflammatory processes.45 The reason why tenascin-C was transiently expressed in the laser-induced model may be that laser-induced CNV is largest by 7 days after photocoagulation and spontaneously diminishes thereafter in this model.27 In contrast, in AMD patients, our immunofluorescent staining and the findings of previous studies demonstrated that tenascin-C is expressed by transdifferentiated RPE cells (myofibroblasts) at the RPE-choroid interface during the active phase of CNV membrane formation, but absent in old quiescent scars.24, 46 Our finding is consistent with those of previous studies showing that in patients with chronic inflammatory diseases such as keloids and sclerodermas, tenascin-C is expressed by fibroblasts.47, 48 Therefore, it is likely that tenascin-C is continuously produced by myofibroblasts at the active edge of CNV in AMD patients and is not a transient phenomenon that peaks at 3 days.
The results of this study indicated that tenascin-C siRNA inhibited the formation of CNV in vitro and in vivo. Although aflibercept, a VEGF trap, is most commonly used for neovascular AMD (based on the clinical evidence),6, 49 a recent report has suggested that the Fc portion of anti-VEGF drugs facilitates RPE cells transdifferentiation.50 Our study demonstrated that transdifferentiation of hRPE cells into myofibroblasts results in the production of tenascin-C. These results suggest that aflibercept promotes CNV development via tenascin-C by facilitating RPE cell transdifferentiation. Therefore, aflibercept has a dual and conflicting mechanism of action; on one hand, it has anti-VEGF properties that inhibit CNV formation, and on the other hand, it may have pro-angiogenic potential induced by tenascin-C from transdifferentiated RPE cells. These opposing effects may partially explain the tolerance or tachyphylaxis developed after long-term treatment with anti-VEGF drugs. Therefore, a combination of aflibercept and tenascin-C siRNA could exert a more potent effect on the eyes with neovascular AMD, compared with singular aflibercept therapy.
In summary, we discovered that tenascin-C secreted from transdifferentiated RPE cells promotes CNV formation in vitro via integrin αVβ3, in a paracrine fashion. Furthermore, tenascin-C knockout mice and mice that received a vitreous injection of tenascin-C siRNA showed a significant reduction in CNV volume. Therefore, tenascin-C could be an effective therapeutic target for combating AMD-associated CNV.
References
Lim LS, Mitchell P, Seddon JM et al, Age-related macular degeneration. Lancet 2012; 379: 1728–1738.
Campochiaro PA, Soloway P, Ryan SJ et al, The pathogenesis of choroidal neovascularization in patients with age-related macular degeneration. Mol Vis 1999; 5: 34.
Rosenfeld PJ, Brown DM, Heier JS et al, Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355: 1419–1431.
Group CR, Martin DF, Maguire MG et al, Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011; 364: 1897–1908.
Rofagha S, Bhisitkul RB, Boyer DS et al, Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 2013; 120: 2292–2299.
Schmidt-Erfurth U, Kaiser PK, Korobelnik JF et al, Intravitreal aflibercept injection for neovascular age-related macular degeneration: ninety-six-week results of the VIEW studies. Ophthalmology 2014; 121: 193–201.
Schaal S, Kaplan HJ, Tezel TH . Is there tachyphylaxis to intravitreal anti-vascular endothelial growth factor pharmacotherapy in age-related macular degeneration? Ophthalmology 2008; 115: 2199–2205.
Binder S . Loss of reactivity in intravitreal anti-VEGF therapy: tachyphylaxis or tolerance? Br J Ophthalmol 2012; 96: 1–2.
Gasperini JL, Fawzi AA, Khondkaryan A et al, Bevacizumab and ranibizumab tachyphylaxis in the treatment of choroidal neovascularisation. Br J Ophthalmol 2012; 96: 14–20.
Grunwald JE, Daniel E, Huang J et al, Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology 2014; 121: 150–161.
Castellon R, Caballero S, Hamdi HK et al, Effects of tenascin-C on normal and diabetic retinal endothelial cells in culture. Invest Ophthalmol Vis Sci 2002; 43: 2758–2766.
Ishikawa K, Yoshida S, Kobayashi Y et al, Microarray analysis of gene expression in fibrovascular membranes excised from patients with proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 2015; 56: 932–946.
Kobayashi Y, Yoshida S, Zhou Y et al, Tenascin-C promotes angiogenesis in fibrovascular membranes in eyes with proliferative diabetic retinopathy. Mol Vis 2016; 22: 436–445.
Orend G, Chiquet-Ehrismann R . Tenascin-C induced signaling in cancer. Cancer Lett 2006; 244: 143–163.
Chiquet-Ehrismann R, Chiquet M . Tenascins: regulation and putative functions during pathological stress. J Pathol 2003; 200: 488–499.
Willems IE, Arends JW, Daemen MJ . Tenascin and fibronectin expression in healing human myocardial scars. J Pathol 1996; 179: 321–325.
Wallner K, Sharifi BG, Shah PK et al, Adventitial remodeling after angioplasty is associated with expression of tenascin mRNA by adventitial myofibroblasts. J Am Coll Cardiol 2001; 37: 655–661.
Taki J, Inaki A, Wakabayashi H et al, Dynamic expression of tenascin-C after myocardial ischemia and reperfusion: assessment by 125I-anti-tenascin-C antibody imaging. J Nucl Med 2010; 51: 1116–1122.
Garcion E, Faissner A, ffrench-Constant C . Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 2001; 128: 2485–2496.
Tanaka K, Hiraiwa N, Hashimoto H et al, Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int J Cancer 2004; 108: 31–40.
Calvo A, Catena R, Noble MS et al, Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene 2008; 27: 5373–5384.
Pezzolo A, Parodi F, Marimpietri D et al, Oct-4+/tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res 2011; 21: 1470–1486.
Grossniklaus HE, Green WR . Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Submacular Surgery Trials Research Group. Arch Ophthalmol 1998; 116: 745–749.
Nicolo M, Piccolino FC, Zardi L et al, Detection of tenascin-C in surgically excised choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol 2000; 238: 107–111.
Nakama T, Yoshida S, Ishikawa K et al, Inhibition of choroidal fibrovascular membrane formation by new class of RNA interference therapeutic agent targeting periostin. Gene Ther 2015; 22: 127–137.
Chang B, Hawes NL, Hurd RE et al, Retinal degeneration mutants in the mouse. Vision Res 2002; 42: 517–525.
Lambert V, Lecomte J, Hansen S et al, Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc 2013; 8: 2197–2211.
Ishikawa K, Yoshida S, Nakao S et al, Bone marrow-derived monocyte lineage cells recruited by MIP-1beta promote physiological revascularization in mouse model of oxygen-induced retinopathy. Lab Invest 2012; 92: 91–101.
Kobayashi Y, Yoshida S, Nakama T et al, Overexpression of CD163 in vitreous and fibrovascular membranes of patients with proliferative diabetic retinopathy: possible involvement of periostin. Br J Ophthalmol 2015; 99: 451–456.
Yoshida S, Ishikawa K, Asato R et al, Increased expression of periostin in vitreous and fibrovascular membranes obtained from patients with proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 2011; 52: 5670–5678.
Jin M, He S, Worpel V et al, Promotion of adhesion and migration of RPE cells to provisional extracellular matrices by TNF-alpha. Invest Ophthalmol Vis Sci 2000; 41: 4324–4332.
Ishikawa K, Yoshida S, Nakao S et al, Periostin promotes the generation of fibrous membranes in proliferative vitreoretinopathy. FASEB J 2014; 28: 131–142.
Amin R, Puklin JE, Frank RN . Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest Ophthalmol Vis Sci 1994; 35: 3178–3188.
Hanamura N, Yoshida T, Matsumoto E et al, Expression of fibronectin and tenascin-C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int J Cancer 1997; 73: 10–15.
Imanaka-Yoshida K, Hiroe M, Nishikawa T et al, Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest 2001; 81: 1015–1024.
El-Karef A, Kaito M, Tanaka H et al, Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J Hepatol 2007; 46: 664–673.
Pfeffer BA, Flanders KC, Guerin CJ et al, Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp Eye Res 1994; 59: 323–333.
Yuan F, Fu X, Shi H et al, Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PLoS One 2014; 9: e107063.
Cao X, Shen D, Patel MM et al, Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int 2011; 61: 528–535.
Gamulescu MA, Chen Y, He S et al, Transforming growth factor beta2-induced myofibroblastic differentiation of human retinal pigment epithelial cells: regulation by extracellular matrix proteins and hepatocyte growth factor. Exp Eye Res 2006; 83: 212–222.
Ishikawa K, Kannan R, Hinton DR . Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp Eye Res 2015; 142: 19–25.
Kent D, Sheridan C . Choroidal neovascularization: a wound healing perspective. Mol Vis 2003; 9: 747–755.
Friedlander M, Theesfeld CL, Sugita M et al, Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci USA 1996; 93: 9764–9769.
Sriramarao P, Mendler M, Bourdon MA . Endothelial cell attachment and spreading on human tenascin is mediated by alpha 2 beta 1 and alpha v beta 3 integrins. J Cell Sci 1993; 105 (Pt 4): 1001–1012.
Johnson LV, Leitner WP, Staples MK et al, Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res 2001; 73: 887–896.
Fasler-Kan E, Wunderlich K, Hildebrand P et al, Activated STAT 3 in choroidal neovascular membranes of patients with age-related macular degeneration. Ophthalmologica 2005; 219: 214–221.
Dalkowski A, Schuppan D, Orfanos CE et al, Increased expression of tenascin C by keloids in vivo and in vitro. Br J Dermatol 1999; 141: 50–56.
Lacour JP, Vitetta A, Chiquet-Ehrismann R et al, Increased expression of tenascin in the dermis in scleroderma. Br J Dermatol 1992; 127: 328–334.
Kumar N, Marsiglia M, Mrejen S et al, Visual and anatomical outcomes of intravitreal aflibercept in eyes with persistent subfoveal fluid despite previous treatments with ranibizumab in patients with neovascular age-related macular degeneration. Retina 2013; 33: 1605–1612.
Chen CL, Liang CM, Chen YH et al, Bevacizumab modulates epithelial-to-mesenchymal transition in the retinal pigment epithelial cells via connective tissue growth factor up-regulation. Acta Ophthalmol 2012; 90: e389–e398.
Acknowledgements
The authors would like to thank Masayo Eto for the technical assistance provided. This study was supported by Grants-in-Aid for Scientific Research (B) (Nos. 15H04995 and 26293374), a Grant-in-Aid for Challenging Exploratory Research (No. 26670757), and a Grant-in-Aid for JSPS Fellows from Japan Society for the Promotion of Science (No. 15J03433).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Rights and permissions
About this article

Cite this article
Kobayashi, Y., Yoshida, S., Zhou, Y. et al. Tenascin-C secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin αV. Lab Invest 96, 1178–1188 (2016). https://doi.org/10.1038/labinvest.2016.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.99
This article is cited by
-
Increased expression of periostin and tenascin-C in eyes with neovascular glaucoma secondary to PDR
Graefe's Archive for Clinical and Experimental Ophthalmology (2020)
-
Periostin in vitreoretinal diseases
Cellular and Molecular Life Sciences (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}