
Abstract
Hereditary spastic paraplegia (HSP) is a neurological disorder characterized by a progressive spasticity and muscle weakness of the lower limbs. It is divided into two subtypes, uncomplicated and complicated forms. Biallelic mutations in the cytochrome P450 2U1 gene (CYP2U1) are associated with spastic paraplegia type 56 (SPG56), manifesting both uncomplicated and complicated HSP. Accompanying clinical features include intellectual disability, dystonia, cerebellar ataxia, subclinical peripheral neuropathy, visual impairment, as well as abnormalities in brain magnetic resonance imaging. As a rare clinical feature, delayed myelination has previously been reported in only two patients with CYP2U1 mutations. Here, we report a patient with SPG56 with novel compound heterozygous mutations in CYP2U1 which were identified by whole exome sequencing. Our patient exhibited complex features together with delayed myelination, broadening the phenotypic spectrum of SPG56, and implying that CYP2U1 should be screened in HSP with delayed myelination.
Similar content being viewed by others


Introduction
Hereditary spastic paraplegia (HSP) is a neurological disease characterized by a progressive spasticity and lower-limb muscle weakness.1 Modes of HSP inheritance include autosomal dominant, autosomal recessive and X-linked recessive, as well as de novo occurrence. HSP is divided into two subtypes: uncomplicated and complicated.1, 2, 3 Uncomplicated HSP presents with progressive bilateral leg spasticity and weakness, while complicated forms show additional neurological or extra-neurological features.
To date, 69 genes and/or loci are registered in OMIM (https://www.omim.org/phenotypicSeries/PS303350) for HSP. Among them, the cytochrome P450 2U1 gene (CYP2U1), encoding a member of the cytochrome P450 superfamily of enzymes, is associated with autosomal recessive spastic paraplegia type 56 (SPG56) (MIM 615030), also known as SPG49 in HUGO Nomenclature Committee. Patients with CYP2U1 mutations mainly have complicated HSP, although some cases are uncomplicated. Previously reported additional features include intellectual disability, cerebellar ataxia, dystonia, subclinical peripheral neuropathy and optic atrophy.4, 5, 6, 7, 8 Rarely, a thin corpus callosum, basal ganglia calcification and periventricular white matter lesions or delayed myelination have been found by brain magnetic resonance imaging (MRI).4, 6, 7, 8 Here, we report a Japanese patient with SPG56 having novel compound heterozygous mutations in CYP2U1. This is the first report of a Japanese patient with SPG56 including delayed myelination, extending our understanding of the clinical features of SPG56/CYP2U1-related HSP.
Case report
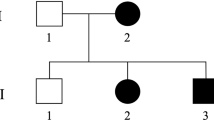
The patient was a 4-year-old boy at the time of the study. He is the third child of healthy, non-consanguineous Japanese parents (Figure 1a), and has a healthy elder brother and a sister. There are no other affected family members. His birth was uneventful. He attained head control at 4 months of age, rolled over at 6 months, crawled at 9 months of age without alternate movement of the legs, and spoke several words at 3 years of age. However, he was not able to stand, walk or use a pincer grasp at 4 years.

Familial pedigree and CYP2U1 mutations. (a) Familial pedigree. (b) Electropherograms of identified CYP2U1 mutations. c.1055C>T was inherited from the father, while c.1616T>G was inherited from the mother. (c) The evolutionary conservation of mutated amino acids (Ala352 and Ile539) from Danio rerio to Homo sapiens. (d) Schematic of CYP2U1 and its mutations. Upper, novel mutations identified in our study; lower: previously described mutations. Blue boxes indicate coding exons, and light blue boxes indicate untranslated regions. A full color version of this figure is available at the Journal of Human Genetics journal online.
At the age of 1, he visited a hospital for developmental delay. On neurological examination, he showed muscle weakness and spasticity of both lower and upper limbs. Contracture of the ankles was also observed. Deep tendon reflexes in the lower and right upper limbs were increased with a positive forced grasp, and the palmomental reflex and Babinski sign were noted. No involuntary movements or cerebellar ataxia were observed. There is no foot deformity such as a cavovarus foot. Blood chemistries including activities of the lysosomal enzymes β-galactosidase, β-hexosaminidase, arylsulfatase and galactocerebrosidase were all normal. The plasma concentration of very long chain fatty acids was not elevated. A nerve conduction study in tibial nerves revealed that motor conduction velocities and compound muscle action potentials were within normal ranges (Supplementary Figure 1).
At 13 months and 26 months of age, brain MRI revealed delayed myelination in the frontal, temporal, parietal and occipital lobes, and insula, and minimal progress of myelination during this interval without demyelination (Figures 2a and b). Thinning of the corpus callosum was not observed (Figure 2c). Developmental evaluation at 3 years and 10 months of age (using the Kyoto Scale of Psychological Development), indicated a total developmental quotient of 26 (equivalent to that of 12 months) with subcategorized quotients as follows: Postural-Motor 11 (equivalent to 5 months), Cognitive-Adaptive 30 (equivalent to 14 months) and Language-Social 29 (equivalent to 13 months).

Brain magnetic resonance imaging (MRI) of the patient. Axial T2 weighted images of the brain MRI at 12 months (a) and 26 months (b). Both showed delayed myelination in the frontal, temporal, parietal and occipital lobes, and insula, and minimal progress of myelination during this interval without demyelination. Thinning of the corpus callosum was not observed in a sagittal T1 weighted image at 26 months (c).
Genetic analysis
The institutional review board of Yokohama City University School of Medicine approved this study. After obtaining informed consent from the patient’s family, we performed whole exome sequencing as previously described.9 The mean depth of the sequenced region was 108 × , with 92.6% of total coding sequences covered by ⩾20 reads. Two novel heterozygous missense variants in CYP2U1 (NM_183075.2) were detected: c.1055C>T (p.Ala352Val) and c.1616T>G (p.Ile539Arg). Sanger sequencing using DNA from the proband and his parents confirmed that these variants were compound heterozygous (Figure 1b). They were absent in dbSNP138, our 575 normal Japanese in-house exomes, the Exome Aggregation Consortium database, the NHLBI Exome Variant Server (ESP6500) (http://evs.gs.washington.edu/EVS/), and the Human Genetic Variation Browser (http://www.genome.med.kyoto-u.ac.jp/SnpDB/).
Ala352 is conserved in the eukaryotic CYP2U1 protein, and Ile539 is conserved among several species including Homo sapiens, Bos taurus and Xenopus tropicalis (Figure 1c). Possible pathogenic effects of these two variants on protein function were evaluated by web-based tools SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/). The c.1055C>T variant was predicted to be pathogenic by all three tools, while the c.1616T>G variant was predicted to be pathogenic by SIFT.
Discussion
To date, a total of 11 CYP2U1 mutations have been reported in 18 patients from nine families with SPG56/CYP2U1-related HSP (Figure 1d). Their clinical presentations are summarized in Table 1. Clinically, they often presented with complex forms of HSP, accompanied by various clinical features. Our patient showed delayed myelination, which is a rare feature of HSP as seen in spastic paraplegia 2 (MIM 312920) by proteolipid mutations and spastic paraplegia 44 by GJC2 (MIM 613206), and the third known case out of ten pedigrees with SPG56/CYP2U1-mutated HSP. Therefore, delayed myelination can be recognized as a possible clinical feature for SPG56, and CYP2U1 screening should be considered in patients with HSP accompanying delayed myelination.
It is thought that the patho-mechanism of HSP includes defects in axon transportation, cytoskeleton control, mitochondrial function and/or axon development.10 Regarding SPG56/CYP2U1-related HSP, mitochondrial dysfunction is suspected as a disease-causing mechanism. CYP2U1 is one of the most conserved cytochrome P450 proteins that catalyze the hydroxylation of arachidonic acid and related fatty acids. Its metabolites are local mediators of signal transduction,4 and might indirectly mediate mitochondrial function via receptors in the brain that regulate mitochondrial respiration and energy production.11 CYP2U1 was also shown to be partially localized in mitochondria, and mutations in CYP2U1 might cause structural abnormalities in mitochondrial membranes, leading to the dysregulation of mitochondrial dynamics and respiratory impairment.4, 11 Indeed, CYP2U1-regulated mitochondrial function was preciously shown to be important for neuronal function in the corticospinal tract.4 Our report, together with two other previous cases of delayed myelination in cerebral white matter,8 further supports a possible association between normal myelination and mitochondrial function.
References
Fink, J. K. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328 (2013).
Depienne, C., Stevanin, G., Brice, A. & Durr, A. Hereditary spastic paraplegias: an update. Curr. Opin. Neurol. 20, 674–680 (2007).
Fink, J. K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 6, 65–76 (2006).
Tesson, C., Nawara, M., Salih, M. A., Rossignol, R., Zaki, M. S., Al Balwi, M. et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 91, 1051–1064 (2012).
Citterio, A., Arnoldi, A., Panzeri, E., D'Angelo, M. G., Filosto, M., Dilena, R. et al. Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J. Neurol. 261, 373–381 (2014).
Leonardi, L., Ziccardi, L., Marcotulli, C., Rubegni, A., Longobardi, A., Serrao, M. et al. Pigmentary degenerative maculopathy as prominent phenotype in an Italian SPG56/CYP2U1 family. J. Neurol. 263, 781–783 (2016).
Masciullo, M., Tessa, A., Perazza, S., Santorelli, F. M., Perna, A. & Silvestri, G. Hereditary spastic paraplegia: novel mutations and expansion of the phenotype variability in SPG56. Eur. J. Paediatr. Neurol. 20, 444–448 (2016).
Kariminejad, A., Schols, L., Schule, R., Tonekaboni, S. H., Abolhassani, A., Fadaee, M. et al. CYP2U1 mutations in two Iranian patients with activity induced dystonia, motor regression and spastic paraplegia. Eur. J. Paediatr. Neurol. 20, 782–787 (2016).
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 (2013).
Salinas, S., Proukakis, C., Crosby, A. & Warner, T. T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138 (2008).
Benard, G. & Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 20, 365–374 (2009).
Acknowledgements
We thank all of the participants for their cooperation in this research. We are also grateful to Mr T. Miyama, Ms K. Takabe, Ms N. Watanabe, Ms S. Sugimoto, Mr S Nakamura and Ms M. Sato from the Department of Human Genetics, Yokohama City University Graduate School of Medicine for their technical assistance. This work was supported by grants from Research on Measures for Intractable Diseases (NM), Comprehensive Research on Disability Health and Welfare (NM), the Strategic Research Program for Brain Science (NM), the Initiative on Rare and Undiagnosed Diseases in Pediatrics (NM) and the Initiative on Rare and Undiagnosed Diseases in Adults (NM), from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants-in-Aid for Scientific Research (A (NM), B (NM, HS) and C (SM)) from the Japan Society for the Promotion of Science; the fund for the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (NM) from the Japanese Science and Technology Agency; grants from the Ministry of Health, Labor and Welfare (NM); the Takeda Science Foundation (NM, HS, NM); and The Ichiro Kanehara Foundation for the Promotion of Medical Science & Medical Care (SM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Authors declare noconflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Minase, G., Miyatake, S., Nabatame, S. et al. An atypical case of SPG56/CYP2U1-related spastic paraplegia presenting with delayed myelination. J Hum Genet 62, 997–1000 (2017). https://doi.org/10.1038/jhg.2017.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.77
This article is cited by
-
Spastic Paraplegia Type 56 in a Young Child
The Indian Journal of Pediatrics (2020)
