
Abstract
Alzheimer’s disease (AD) and frontotemporal dementia (FTD) are two common forms of primary neurodegenerative dementia that show overlapping clinical symptoms. The aim of this study was to perform genetic analyses on GRN, VCP, CHMP2B, FUS, TARDBP, C9orf72 and MAPT genes in Chinese AD and FTD patients. We performed gene sequencing of the GRN, VCP, CHMP2B, FUS, TARDBP, MAPT and C9orf72 genes in 61 clinical AD and 38 FTD Chinese patients. We identified a known mutation of MAPT (p.Pro301Leu, c.902C>T) in four patients from an autosomal dominant FTD family with behavioral variant FTD (bvFTD) and progressive nonfluent aphasia (PNFA) phenotypes, and a novel mutation in MAPT (p.Leu48Val, c.142 G>C) in a sporadic progressive supranuclear palsy patient. Two novel variations in VCP (p.Thr127Ala, c. 379A>G; p.Asn401Ser, c.1202A>G) were present in both a sporadic FTD and an AD case, and a novel deletion in GRN (560del p.Leufs) was found in a sporadic primary progressive aphasia patient. Mutations of VCP, GRN and MAPT genes are present in Chinese FTD cases. In the case of the MAPT mutation, the family presented with both bvFTD and PNFA phenotypes, while the VCP mutation was also related to an early-onset AD phenotype.
Similar content being viewed by others

Introduction
Frontotemporal dementia (FTD) represents a group of clinically, neuropathologically and genetically heterogeneous disorders. Presenting as a range of progressive dementia syndromes associated with the focal atrophy of the orbitomedial frontal and anterior temporal lobes, FTD often begins in the patient’s fifth to seventh decade of life. Epidemiological studies suggest that in patients younger than 65 years, the incidence and prevalence of FTD is similar to that of AD.1 FTD can be classified into two main cognitive syndromes:2 behavioral variant FTD (bvFTD) and primary progressive aphasia (PPA), whose diagnostic criteria have been recently revised to include neuroimaging and genetics testing.3, 4 In addition, FTD overlaps with motor neuron disease or amyotrophic lateral sclerosis (FTD-MND or FTD-ALS), as well as the parkinsonian syndromes, progressive supranuclear palsy and corticobasal syndrome.
Familial aggregation is frequently reported in FTD, with about 10% of cases showing an autosomal dominant transmission. Genes demonstrated to be responsible for familial FTD include those for: the microtubule-associated protein tau (MAPT), progranulin (GRN), valosin-containing protein (VCP), chromatin-modifying protein 2B (CHMP2B), the FUS protein (FUS), the TAR-DNA-binding protein 43 encoding gene (TARBDP), and, very recently, a novel hexanucleotide expansion in chromosome 9 (C9orf72).5, 6 Interestingly, GRN and MAPT have been found associated with clinically diagnosed AD cases.7, 8, 9, 10, 11, 12, 13, 14
Because of the advanced developments in next-generation sequencing, the high-throughput sequencing of targeted genomic regions of many individuals in a single run is now a cheap and feasible technology for medical research. In this study, we utilized next-generation sequencing to detect gene variants in MAPT, GRN, VCP, FUS, TARBDP and CHMP2B, and used a repeat-primed polymerase chain reaction method to screen for C9orf72 in 61 clinically probable AD and 38 FTD patients seen at Tianjin Huanhu Hospital. All such patients had experienced their first symptoms of dementia before the age of 70. Clinical findings and neuroimages were analyzed in patients with FTD-related gene mutations in this study.
Materials and methods
Subjects
The cohort in this study consisted of 61 AD and 38 FTD patients (37.4% males and 62.6% females) from a clinical case series, diagnosed in the Department of Neurology at Tianjin Huanhu Hospital between 2009 and 2013. All individuals in this study were of Chinese Han ethnicity. The criteria of the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) were used for the diagnosis of probable AD,15 and Diagnostic and Statistical Manual of Mental Disorders—fourth edition, Text Revision (DSM-IV-TR) criteria16 were used for the diagnosis of dementia. A diagnosis of probable FTD was established according to the criteria published in 2011.3, 4 The onset of dementia in all of these patients occurred before 70 years of age, with a mean age of onset of 60.5±6.1 years (range 40–69 years). In 78 patients (78.8% of the cohort), the onset of dementia occurred before the age of 65. A positive family history, defined as having at least one affected first- or second-degree relative with dementia, was found in 26 patients (26.3%). Four patients came from one family. None of the other patients or controls were related. For genetic studies, a total of 146 healthy control subjects were also studied. All subjects agreed to participate in the study, and biological samples were obtained after written, informed consent. The study was approved by the Huanhu Hospital Ethics Committee.
Gene sequencing
Gene selection
A total of 89 candidate genes were selected for sequence analysis. Six genes associated with FTD, which consisted of CHMP2B, FUS, GRN, MAPT, TARDBP and VCP, were selected. The ‘AD’ gene group included 83 genes that have mutations known to be associated with AD.
Target region capture and sequencing
Genomic DNA was extracted from peripheral blood according to the manufacturer’s instructions using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Qualified genomic DNA samples were randomly fragmented into fragments with base pair peaks of 200–300 bp. DNA fragments were then amplified, purified and hybridized with probes using a custom capture array (NimbleGen; Roche, Mannheim, Germany). Each captured library was subjected to massive parallel sequencing using a 91 paired-end protocol using an Illumina HiSeq 2000 Analyzer (San Diego, CA, USA).
Data analysis and variant calling
Bioinformatics analysis was initiated from sequencing data (raw data), which were generated from an Illumina pipeline. A few sequences were removed from the primary data using a local dynamic programming algorithm, including low-quality reads (<Q10). The remaining ‘clean data’ were aligned to the human reference genome (UCSC build hg19, 200902 release, http://genome.ucsc.edu/) using the Burrows-Wheeler Aligner software (BWA) (Wellcome Trust Sanger Institute, Wellcome Genome Campus, Cambridge, UK). Single nucleotide polymorphisms and small insertion/deletions (INDELS) were detected using SOAPsnp software (BGI, Shenzhen, China) and SAMtools (GitHub, San Francisco, CA, USA), respectively. All single nucleotide polymorphisms were determined using the NCBI dbSNP137, HapMap, 1000 human genome data set (20110521 release, http://www.1000genomes.org/). All rare (minor allele frequency of <1%), novel and potentially functional variants, or previously identified pathogenic variants in CHMP2B, FUS, GRN, MAPT, TARDBP and VCP genes were selected for direct sequencing using an ABI PRISM 3130 automated sequencer (PE Applied Biosystems, Foster City, CA, USA) and standard protocols. Primer sequences are available on request. For the identification of novel nucleotide changes, all 146 control DNAs were analyzed at the same time.
C9orf72 repeat expansions
We screened for the presence of the GGGGCC hexanucleotide expansion of C9orf72 using a two-step polymerase chain reaction protocol.17 A cutoff value of 30 repeats was used to define the pathogenic threshold.6
Results
Mutation analyses of FTD genes
A total of one known and one novel missense mutation in MAPT, a novel variant in VCP and a novel deletion in GRN were identified in seven individuals (18.4%) from four unrelated FTD or progressive supranuclear palsy families. A novel variant in VCP was identified in a sporadic patient with an AD phenotype. No other CHMP2B, FUS, GRN, MAPT, TARDBP or VCP mutations were identified in the cases or controls. No pathogenic repeats (>30 repeats) in C9orf72 were detected in these patients or controls. The mutation of genes related to AD has been previously reported elsewhere.18
The known MAPT gene missense mutation (p.Pro301Leu, c.902C>T, exon10, R2) was present in an early-onset FTD family, including four FTD patients. A novel mutation in MAPT (p.Leu48Val, c.142 G>C, exon 2, N-terminal) was found in a sporadic progressive supranuclear palsy patient. The first novel variation in VCP (p.Thr127Ala, c. 379A>G, exon 4, CDC48) was found in a sporadic FTD case. A second novel mutation in VCP (p.Asn401Ser, c.1202A>G, exon11, D1) was found in a sporadic AD case. A novel deletion in GRN (p.Leu187fs, c.560delT, exon 6, Inter FB) was found in a sporadic PPA case.
Clinical characteristics of mutation carriers
Clinical characteristics of the eight clinical probable AD or FTD patients with pathogenic mutations identified in this study are summarized in Table 1.
A pedigree with a three-generation history of early-onset dementia of the FTD type was identified associated with a known mutation of MAPT (p.Pro301Leu). Eight individuals in this pedigree have been diagnosed with a dementia disorder. The transmission of FTD in this family is consistent with an autosomal dominant inheritance (Figure 1).

Family 1 with a MAPT Pro301Leu mutation. Age at death, or current age and age of disease onset (in parentheses) are indicated. A full color version of this figure is available at the Journal of Human Genetics journal online.
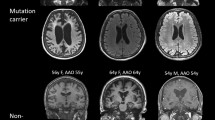
The proband (III-4), a 58-year-old female, developed a progressive language disorder with gradual onset at 54 years of age. At the onset of disease, she experienced difficulty naming familiar objects, and forgot the name of familiar friends and relatives, but did not display comprehension deficits. At 56 years of age, she became increasingly forgetful. At age 58, she exhibited significant cognitive impairment, with a Mini-Mental State Examination (MMSE) score of 24/30 and a Montreal Cognitive Assessment (MoCA) score of 14/30. Her Boston Naming Test score was 4/15. Magnetic resonance imaging (MRI) of her head at 58 years of age showed left temporal lobe atrophy. Fluorodeoxyglucose (FDG) positron emission tomography (PET) scans found hypometabolism in the bilateral temporal cortex (left>right), frontal cortex, anterior cingulate cortex, thalamus, insula and caudate nucleus. A Pittsburgh compound B (PIB)-PET scan did not show cortical amyloid deposits (Figure 2a).

Fluorodeoxyglucose (FDG) positron emission tomography (PET) and Pittsburgh compound B (PIB) PET. (a) The proband of family 1 with a MAPT Pro301Leu mutation, for whom FDG PET scans showed hypometabolism in the bilateral temporal cortex (left>right), frontal cortex, anterior cingulate cortex, thalamus, insula and caudate nucleus; (b) A progressive supranuclear palsy (PSP) patient with a MAPT Leu48Val mutation, for whom FDG PET showed hypometabolism in the bilateral temporal cortex, insula, thalamus, lenticular nucleus, caudate nucleus and midbrain; (c) A patient with a VCP Thr127Ala mutation, who showed hypometabolism in the bilateral frontal lobe, insular lobe, cingulate syrus, caudate nucleus, right lateral temporal lobe and thalamus. Cortical amyloid deposits were not evident on PIB PET in three patients. SPM, Statistical Parametric Mapping.
The III-2, a 62-year-old female, developed progressive language impairment and behavioral changes from 59 years of age. Neuropsychological testing at 62 years of age revealed a MMSE score of 4/30 and a MoCA score of 1/30, with orientation partially reserved. A brain MRI undertaken at 62 years of age showed bilateral frontal and temporal lobe atrophy (Figure 3a). The III-3, a 59-year-old female, developed progressive behavioral changes from 54 years of age. She has been cared for by her family since she was 58. Neuropsychological testing at 59 years of age revealed a MMSE score of 1/30 and a MoCA score of 0/30. The III-5, 56-year-old male, had developed a right hand tremor, and memory loss from 53 and 55 years of age, respectively. Neurological examination showed a postural tremor of the right hand. Neuropsychological testing at this time revealed a MMSE score of 20/30 and a MoCA score of 14/30. The patient’s activities of daily living score was 22. MRI showed mild left frontal and temporal lobe atrophy (Figure 3b). The patient was diagnosed with atypical FTD. The mother of III-2, III-3, III-4 and III-5 showed behavioral changes from 45 years of age and was diagnosed with a mental illness. From 50 years of age, the mother showed memory loss and subsequently died at 54. Two male siblings of the mother developed dementia before 50 years of age, and died at 54 and 56, respectively. Their grandfather died at 60 with dementia and had a mental illness from the age of 50. An older brother (III-1; 66 years of age), who is also a MAPT (p.Pro301Leu) mutation carrier, has shown a decline in memory for a year; MMSE and MoCA scores were both 29/30 and his activities of daily living score was 20. A head MRI appeared normal.

Head magnetic resonance images (MRI) on coronal section. (a) Family 1–3 with a MAPT Pro301Leu mutation, and bilateral frontal and temporal lobe atrophy; (b) Family 1–4 with mild left frontal and temporal lobe atrophy; (c) A patient with a VCP p.Asn401Ser mutation, and bilateral parietal and temporal lobe atrophy (right>left); (d) A patient with a GRN p.Leu187fs mutation, and left frontal and temporal lobe atrophy.
Another 61-year-old female with a MAPT p.Leu48Val mutation had a progressive gait disturbance and dysphagia from when she was 59. At 60 years of age, she became increasingly forgetful. A physical examination revealed poor postural reflexes, axial rigidity, and mild dysarthria, slow vertical saccades, and downgaze palsy. Neuropsychological testing revealed a MMSE score of 22/30 and a MoCA score of 15/30. An MRI of her head showed diffuse cortical and midbrain atrophy, a bilateral temporal lobe atrophy and an enlarged ventricle. FDG PET scans identified hypometabolism in the bilateral temporal cortex, insula, thalamus, lenticular nucleus, caudate nucleus and midbrain. A PIB PET scan did not show cortical amyloid deposits (Figure 2b).
A 61-year-old female patient with a VCP p.Thr127Ala mutation underwent behavioral change and progressive language disturbances from when she was 59. She had lost interest in her appearance and was overeating. She also showed rigidity and bradykinesia, but did not display muscle weakness and atrophy or bone aches. She exhibited significant cognitive impairment, with a MMSE score of 9/30, and her MoCA score was only 2/30. A brain MRI revealed mild atrophy of the bilateral temporal lobe (with the right more obviously affected). An FDG PET scan revealed hypometabolism in the bilateral frontal cortex, insular lobe, cingulate gyrus, caudate nucleus, right lateral temporal lobe and thalamus. A PIB PET scan did not show cortical amyloid deposits (Figure 2c). A 63-year-old female patient with a VCP p.Asn401Ser mutation had progressive memory loss from when she was 61. She also had difficulty in formulating words from 62 years of age. She exhibited significant cognitive impairment, with a MMSE score of 11/30; her MoCA score was only 3/30. A brain MRI revealed bilateral parietal and temporal lobe atrophy (right>left; Figure 3c).
Finally, a 67-year-old female with a novel deletion in GRN (p.Leu187fs, c.560delT) showed progressive speech impediments from 64 years of age and behavioral changes from 66 years of age. From 64 years of age, her speech declined and she became increasingly inarticulate, with perseverations, word-finding and naming difficulties. A behavioral change was very obvious from 66 years of age: she indulged in the overconsumption of sweet food, showed a loss of initiative, wandered for many hours outside her home, but could still find her own way back home. On examination, she could not speak or communicate with others. A head MRI showed left frontal and temporal lobe atrophy (Figure 3d).
Discussion
This is one of the few studies, to our knowledge, undertaken to confirm the presence of MAPT, GRN and VCP mutations in dementia cases of Chinese Han ethnicity. We identified a known mutation in MAPT (p.Pro301Leu), a novel variant in MAPT (p.Leu48Val), two novel variants in VCP (p.Thr127Ala, p.Asn401Ser) and a novel deletion in GRN (p.Leu187fs), after screening 61 AD and 38 FTD patients. Seven patients with mutations (18.4%) were part of the cohort of 38 FTD patients, with an onset of dementia prior to 70 years of age. A patient with a VCP mutation showed an AD phenotype. Such novel mutations, however, were not found in the 146 healthy control individuals, strongly indicating that these variants may increase the risk for developing dementia.
We found a known mutation of MAPT in a large family that showed early-onset dementia across three generations. These patients presented with different clinical phenotypes, including bvFTD and PNFA, with the age of dementia onset from 53 to 59 years of age. The dementia phenotype was inherited in a manner consistent with a simple dominant pattern of inheritance, and consistent with other tau mutations causing FTD. One carrier was 66 years old with normal cognition, suggesting the existence of additional factors that modulate this disease phenotype. The clinical presentation of MAPT mutation carriers was mainly consistent with bvFTD, with a mean onset in the patients’ fifties.19, 20 Nevertheless, cases of progressive nonfluent aphasia have been reported as well, with an onset even in the sixth decade of life.20
Two novel variations in VCP were present in a sporadic FTD case and also in an AD case. Mutations in VCP cause inclusion body myopathy associated with Paget's disease of bone, and FTD (IBMPFD). FTD was diagnosed in 30.34% of symptomatic individuals with VCP mutations, and occurred at a mean age of 55.3 years (range 46–79 years). The female to male ratio was ~2:1.21, 22 With regard to the patient with an FTD phenotype, a brain MRI scan showed mild atrophy of the bilateral temporal lobe (with the right more obviously affected). FDG PET revealed hypometabolism in the bilateral frontal cortex, insular lobe, cingulate gyrus, caudate nucleus, right lateral temporal lobe and thalamus. A PIB PET scan showed no PIB deposits. Such findings support a diagnosis of typical FTD. The other patient showed an AD phenotype with memory loss and word-finding difficulties. After 2 years’ follow-up, the patient showed no obvious behavioral changes. Interestingly, a VCP mutation causing atypical IBMPFD has recently been reported in China; however, members of this family were not diagnosed with FTD.23 In our two VCP mutation carriers, both had early-onset dementia, but did not show any muscle weakness and atrophy or bone aches. Kaleem et al.23 performed several genome-wide linkage screens in late-onset AD patients and identified a novel R92H mutation within the VCP gene. An individual with an R155H mutation and two apolipoprotein E (APOE) 4 alleles was diagnosed with AD.24
The GRN mutation patient showed typical progressive primary aphasia, with obvious left frontal and temporal lobe atrophy. Mutations in GRN are associated with extremely heterogeneous FTD phenotypes, in addition to the classical FTD presentations, AD,25 corticobasal syndrome26 or mild cognitive impairment.27 Mutations in GRN are also related to PPA. One report has described two families with a GRN mutation,28 both showing PPA combined with a behavioral disorder. A second study on two families showed most individuals exhibited a PPA phenotype in association with a GRN mutation.29 There has been an attempt to define the clinical characteristics of PPA associated with a GRN mutation.30 Regrettably, however, we did not test serum progranulin levels in the GRN mutation patient identified in our studies.
In China, the most common assessment tools for dementia or mild cognitive impairment are the Mandarin version of the MMSE, the MoCA, the activities of daily living score and the Clinical Dementia Rating (CDR) Scale. The Mandarin version of the MMSE has been used as a cognitive assessment in dementia screening surveys since 1988, and has been commonly used in clinical assessments.31 The MoCA has been more commonly used as a screening tool for mild cognitive impairment,32 vascular cognitive impairment33 and Parkinson's disease dementia.34 A Chinese version of Addenbrooke's Cognitive Examination-Revised has been recently used in the assessment of AD and mild cognitive impairment.35 However, further work still needs to be undertaken in comparing different assessment tools for dementia.
Conclusions
In summary, we have identified MAPT, VCP and GRN mutations in FTD patients. A MAPT mutation in the same family may be manifested by different clinical phenotypes, whereas mutations in VCP can be present in an individual with a clinical presentation indistinguishable from that of typical AD. These results highlight the necessity of screening for both AD and FTD genes, when an autopsy confirmation of diagnosis is unavailable in early-onset dementia patients.
References
Rabinovici, G. D. & Miller, B. L. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24, 375–398 (2010).
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S. et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554 (1998).
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F. et al. Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014 (2011).
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
Lindquist, S. G., Holm, I. E., Schwartz, M., Law, I., Stokholm, J., Batbayli, M. et al. Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur. J. Neurol. 15, 377–385 (2008).
Rademakers, R., Dermaut, B., Peeters, K., Cruts, M., Heutink, P., Goate, A. et al. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum. Mutat. 22, 409–411 (2003).
van Swieten, J. C., Stevens, M., Rosso, S. M., Rizzu, P., Joosse, M., de Koning, I. et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann. Neurol. 46, 617–626 (1999).
Brouwers, N., Nuytemans, K., van der Zee, J., Gijselinck, I., Engelborghs, S., Theuns, J. et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446 (2007).
Kelley, B. J., Haidar, W., Boeve, B. F., Baker, M., Graff-Radford, N. R., Krefft, T. et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol. Aging 30, 739–751 (2009).
Kelley, B. J., Haidar, W., Boeve, B. F., Baker, M., Shiung, M., Knopman, D. S. et al. Alzheimer disease-like phenotype associated with the c.154delA mutation in progranulin. Arch. Neurol. 67, 171–177 (2010).
Rademakers, R., Baker, M., Gass, J., Adamson, J., Huey, E. D., Momeni, P. et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C—>T (Arg493X) mutation: an international initiative. Lancet Neurol. 6, 857–868 (2007).
Yu, C. E., Bird, T. D., Bekris, L. M., Montine, T. J., Leverenz, J. B., Steinbart, E. et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch. Neurol. 67, 161–170 (2010).
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. & Stadlan, E. M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34, 939–944 (1984).
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edn, Text Revision, 157–158 (American Psychiatric Association, Washington, DC, 2000).
Jiao, B., Guo, J. F., Wang, Y. Q., Yan, X. X., Zhou, L., Liu, X. Y. et al. C9orf72 mutation is rare in Alzheimer's disease, Parkinson's disease, and essential tremor in China. Front. Cell Neurosci. 7, 164 (2013).
Shi, Z., Wang, Y., Liu, S., Liu, M., Liu, S., Zhou, Y. et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement. Geriatr. Cogn. Disord. 39, 32–40 (2015).
Yancopoulou, D. & Spillantini, M. G. Tau protein in familial and sporadic diseases. Neuromolecular Med. 4, 37–48 (2003).
Villa, C., Ghezzi, L., Pietroboni, A. M., Fenoglio, C., Cortini, F., Serpente, M. et al. A novel MAPT mutation associated with the clinical phenotype of progressive nonfluent aphasia. J. Alzheimers Dis. 26, 19–26 (2011).
Mehta, S. G., Khare, M., Ramani, R., Watts, G. D., Simon, M., Osann, K. E. et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin. Genet. 83, 422–431 (2013).
Gu, J. M., Ke, Y. H., Yue, H., Liu, Y. J., Zhang, Z., Zhang, H. et al. A novel VCP mutation as the cause of atypical IBMPFD in a Chinese family. Bone 52, 9–16 (2013).
Kaleem, M., Zhao, A., Hamshere, M. & Myers, A. J. Identification of a novel valosin-containing protein polymorphism in late-onset Alzheimer's disease. Neurodegener. Dis. 4, 376–381 (2007).
Mehta, S. G., Watts, G. D., Adamson, J. L., Hutton, M., Umberger, G., Xiong, S. et al. APOE is a potential modifier gene in an autosomal dominant form of frontotemporal dementia (IBMPFD). Genet. Med. 9, 9–13 (2007).
Carecchio, M., Fenoglio, C., De Riz, M., Guidi, I., Comi, C., Cortini, F. et al. Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic mild cognitive impairment converted to Alzheimer's disease. J. Neurol. Sci. 287, 291–293 (2009).
Carecchio, M., Fenoglio, C., Cortini, F., Comi, C., Benussi, L., Ghidoni, R. et al. Cerebrospinal fluid biomarkers in progranulin mutations carriers. J. Alzheimers Dis. 27, 781–790 (2011).
Pietroboni, A. M., Fumagalli, G. G., Ghezzi, L., Fenoglio, C., Cortini, F., Serpente, M. et al. Phenotypic heterogeneity of the GRN Asp22fs mutation in a large Italian kindred. J. Alzheimers Dis. 24, 253–259 (2011).
Snowden, J. S., Pickering-Brown, S. M., Mackenzie, I. R., Richardson, A. M., Varma, A., Neary, D. et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129, 3091–3102 (2006).
Mesulam, M., Johnson, N., Krefft, T. A., Gass, J. M., Cannon, A. D., Adamson, J. L. et al. Progranulin mutations in primary progressive aphasia: the PPA1 and PPA3 families. Arch. Neurol. 64, 43–47 (2007).
Rohrer, J. D., Crutch, S. J., Warrington, E. K. & Warren, J. D. Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia 48, 288–297 (2010).
Katzman, R., Zhang, M. Y., Ouang, Ya, Q., Wang, Z. Y., Liu, W. T., Yu, E. et al. A Chinese version of the Mini-Mental State Examination; impact of illiteracy in a Shanghai dementia survey. J. Clin. Epidemiol. 41, 971–978 (1988).
Yu, J., Li, J. & Huang, X. The Beijing version of the Montreal Cognitive Assessment as a brief screening tool for mild cognitive impairment: a community-based study. BMC Psychiatry 12, 156 (2012).
Tu, Q. Y., Jin, H., Ding, B. R., Yang, X., Lei, Z. H., Bai, S. et al. Reliability, validity, and optimal cutoff score of the Montreal Cognitive Assessment (Changsha version) in ischemic cerebrovascular disease patients of Hunan province, China. Dement. Geriatr. Cogn. Dis. Extra 3, 25–36 (2013).
Chen, L., Yu, C., Fu, X., Liu, W., Hua, P., Zhang, N. et al. Using the Montreal Cognitive Assessment Scale to screen for dementia in Chinese patients with Parkinson's Disease. Shanghai Arch. Psychiatry 25, 296–305 (2013).
Fang, R., Wang, G., Huang, Y., Zhuang, J. P., Tang, H. D., Wang, Y. et al. Validation of the Chinese version of Addenbrooke's cognitive examination-revised for screening mild Alzheimer's disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 37, 223–231 (2014).
Acknowledgements
The study was supported by Tianjin Science and Technology Support Programs (funding numbers: 12ZCZDSY02900 and 13ZCZDSY01600), the Science and Technology Project of Tianjin Municipal Health Bureau (funding number: 2014KR10), and the Tianjin Natural Science Foundation (funding number: 13JCYBJC21300). This manuscript has been edited by native English-speaking experts from BioMed Proofreading LLC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Shi, Z., Liu, S., Xiang, L. et al. Frontotemporal dementia-related gene mutations in clinical dementia patients from a Chinese population. J Hum Genet 61, 1003–1008 (2016). https://doi.org/10.1038/jhg.2016.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.92
This article is cited by
-
Clinical features and biomarkers of semantic variant primary progressive aphasia with MAPT mutation
Alzheimer's Research & Therapy (2023)
-
Recent Advances in the Genetics of Frontotemporal Dementia
Current Genetic Medicine Reports (2019)
