
Abstract
We have used single-nucleotide polymorphism microarray genotyping and homozygosity-by-descent (HBD) mapping followed by Sanger sequencing or whole-exome sequencing (WES) to identify causative mutations in three consanguineous families with intellectual disability (ID) related to thyroid dyshormonogenesis (TDH). One family was found to have a shared HBD region of 12.1 Mb on 8q24.21-q24.23 containing 36 coding genes, including the thyroglobulin gene, TG. Sanger sequencing of TG identified a homozygous nonsense mutation Arg2336*, which segregated with the phenotype in the family. A second family showed several HBD regions, including 6.0 Mb on 2p25.3-p25.2. WES identified a homozygous nonsense mutation, Glu596*, in the thyroid peroxidase gene, TPO. WES of a mother/father/proband trio from a third family revealed a homozygous missense mutation, Arg412His, in TPO. Mutations in TG and TPO are very rarely associated with ID, mainly because TDH is generally detectable and treatable. However, in populations where resources for screening and detection are limited, and especially where consanguineous marriages are common, mutations in genes involved in thyroid function may also be causes of ID, and as TPO and TG mutations are the most common genetic causes of TDH, these are also likely to be relatively common causes of ID.
Similar content being viewed by others


Introduction
Congenital hypothyroidism is the most common endocrine disorder in infants.1 Its prevalence is 1:3000 to 1:4000 births.2 Early detection and treatment can prevent serious neurodevelopmental consequences, including intellectual disability (ID).3 Neonatal screening has been in place in many countries since 1970s.4 Thyroid dyshormonogenesis (TDH; MIM 274400) are inherited defects in hormone biosynthesis, account for 10–15% of all cases of congenital hypothyroidism and can result from disruptions at multiple stages of thyroid hormone biosynthesis. Mutations in seven different genes have been identified to cause TDH. Thyroid peroxidase (TPO) and thyroglobulin (TG)—which provides tyrosyl groups—are the most common causes.5, 6 Many low- and middle-income countries do not have neonatal screening programs for hypothyroidism and congenital hypothyroidism can result in ID.7 In this report, we describe three such families from Pakistan where new mutations in TG and TPO have caused ID.
Materials and methods
Family 1: Ascertainment and mapping of the candidate region has been described previously for this family (Family MR68; Figure 1). This consanguineous family with four affected individuals in two generations was ascertained for the presence of ID from the Dera Ghazi Khan District of Punjab, Pakistan. Written informed consent was taken from all the participants and from their guardians in case of patients. Blood was drawn from the family members and DNA was isolated by standard protocol.9 Patients in this family had a severe form of ID and syndromic features. Patient III-1 has a prominent supraorbital ridge, deep-set eyes and bulbous nasal tip, with mild nasal flaring, thin upper lip and thick lower lip. His ears are mildly protruding and normally formed and set. A prominent goiter is noted. Patient IV-4 has arched thick eyebrows and deep-set eyes. The nose is short with flat nasal root, and wide, bulbous nasal tip with nasal flaring. The philtrum is long and hypoplastic. There is a thin upper lip and thick lower lip and a pointed chin. Patient IV-5 has a short forehead, arched eyebrows, deep-set eyes and infraorbital fullness. The nose is short with flat nasal root and upturned, bulbous nasal tip with nasal flaring. The philtrum is hypoplastic. There is a thick lower lip and goiter is noted.

(a) Family 1 pedigree. Consanguinity between II-1 and II-2 was not documented, but all family marriages were within the clan, and consanguinity is almost certain. Photographs of affected individuals (b) III-1, (c) IV-4 and (d) IV-5. (e) Electropherograms of TG exon 40 showing (top) heterozygote (III-2) at NM_003235.4: c.7006C>T and (bottom) homozygote (individual IV-4) marked by red box. A full color version of this figure is available at the Journal of Human Genetics journal online.
Genotyping with Affymetrix 5.0 (Santa Clara, CA, USA) or 6.0 SNP microarrays was performed. Data were visually inspected by the dChip software (http://biosun1.harvard.edu/complab/dchip),10 and genome-wide homozygosity analyses were performed by HomozygosityMapper (http://www.homozygositymapper.org).11 Additional mapping was performed by using highly polymorphic microsatellite markers (D1S1709, D1S1763E, D1S484, D1S2585, D1S392, D8S490, D8S1207, D8S478, D8S400, D8S346).8 Candidate genes (KCNQ3, LRRC6, TMEM71, ST3GAL1, PHF20L1, ADCY8, TG) were selected for Sanger sequencing from the two homozygosity-by-descent (HBD) regions (~14.3 Mb in total; chr1:159 875 455–161 990 106 and chr8:127 669 856–139 912 945; hg19).
Family 2: this family, in which the parents are first cousins, was recruited through the Lahore Institute of Research and Development. The family is located within Lahore, in Punjab Pakistan and has two affected individuals with mild or moderate ID (see Figure 2). Written informed consent was obtained from all participants and from parents in case of affected individuals. Blood was drawn from family members and DNA was extracted by standard procedures.9 Initial clinical evaluation was performed by a clinical psychologist (MA). Both the patients had mild or moderate form of ID but had no other behavior or psychiatric problem. Patient II-1 with moderate form of ID is noted to have a widow's peak, high forehead, synophrys with medial and lateral flaring of the eyebrows, hooded eyelids, apparent hypertelorism, broad nasal bridge, thick lips, downturned corners of the mouth and large upper incisors. Ears were slightly simple and mildly protruding but normally set and rotated. Patient II-2 with mild form of ID is noted to have a widow's peak, high forehead, thick eyebrows, slightly hooded eyelids, mild apparent hypertelorism, slender nose with prominent nasal tip, thin upper lip and a small, pointed chin with mild retrognathia. Ears were slightly underfolded, normally set and mildly protruding. Both affected individuals (II-1, II-2) were genotyped using Affymetrix 500 K NspI microarrays (Affymetrix; Santa Clara, CA, USA). Homozygosity analysis was performed using FSuite.12 Whole-exome sequencing (WES) was performed for individual II-2 using the SOLiD 5500 sequencing platform, using methodology described previously.13 Sequence alignment and analysis pipeline was also performed as described previously.13

(a) Family 2 pedigree. Photographs of affected individuals (b) II-1, (c) II-2. (d) Electropherograms of TPO exon 11 showing (top) wild type (unrelated control), (middle) heterozygote (I-1) at NM_000547.5:c.1786G>T; and (bottom) homozygote (individual II-1) marked by red box. A full color version of this figure is available at the Journal of Human Genetics journal online.
Family 3: this family, in which the parents are first cousins, was recruited through the Department of Psychiatry, Bolan Medical College, Quetta district of Baluchistan, Pakistan. The parents have two affected offspring, both with moderate form of ID (Figure 3). Written informed consent was obtained from all participants and from parents in case of affected individuals. Blood was drawn from family members and DNA was extracted by standard procedures.9 Both patients had normal births but showed delay in development of motor skills and were able to speak only a few words at the age of 4 years. Both patients have short nose with flat nasal roots and bulbous nasal tip. Apparently both patients have hypertelorism. Both patients frequently have problems with constipation. WES was performed for patient IV:1 and his unaffected parents III-6 and III-7 (Figure 3a), using methods described previously.14

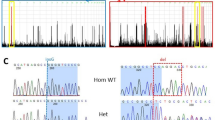
(a) Family 3 pedigree. (b) Electropherograms of TPO exon 8 showing (top) heterozygote (III-6) at NM_000547.5: c.1235G>A (marked by red box), p.Arg412His; homozygous mutation (IV-3) and (bottom) wild-type homozygous individual. Photographs of affected individuals. (c) IV:1, (d) IV-3. (e) Conservation at TPO Arg412His (highlighted in red) shown using Clustal Omega protein sequence alignments (http://www.ebi.ac.uk/Tools/msa/clustalo/) for human TPO (NP_000538.3) against mouse (Mus musculus: NP_033443.1), opossum (Monodelphis domestica; XP_007476256.1), alligator (Alligator Mississippiensis; JH735758.2_prot), frog (Xenopus tropicalis; ENSXETT00000015189.3), zebrafish (Danio rerio; ENSDART00000046464.7), fruit fly (Drosophila melanogaster; NP_523891.2) and nematode (Caenorhabditis elegans; ZK994.3). A full color version of this figure is available at the Journal of Human Genetics journal online.
Results
In this study, three families with TDH and ID were investigated. For Family 1, we found two HBD regions on chromosomes 1 and 8: Chr1:159 875 455–161 990 106 (2.1 Mb; hg19 coordinates) and Chr8:127 669 856–139 912 945 (12.2 Mb).8 Candidate genes were Sanger sequenced. Because of the larger size of HBD region on Chromosome 8 and also the presence of goiter, we prioritized TG and six other genes (KCNQ3, LRRC6, TMEM71, ST3GAL1, PHF20L1, ADCY8) for sequencing. A homozygous nonsense mutation was identified in TG: Chr8:134034365C>T; NM_003235.4: c.7006C>T; p.Arg2336*. This variant segregated within the family in autosomal-recessive manner and is present in the ExAC database (http://exac.broadinstitute.org; accessed December 2015) in 2 alleles (heterozygous) out of 121 412 (rs144875913) but not present in >8000 from South Asian populations and is listed in the Human Gene Mutation Database (HGMD; public version; accessed December 2015) and reported by Machiavelli et al.15 in patient with compound heterozygosity, with a missense variant on the second allele.
HBD analysis of microarray genotype data for Family 2 indicated seven large shared HBD stretches spanning ~48 Mb (Table 1). One 6 Mb region on 2p25.3-p25.2, flanked by single-nucleotide polymorphisms (SNPs) rs4522651 and rs10495544, contains the TPO gene. Whole-exome sequence data analysis for II-2 identified a homozygous nonsense mutation in exon 11 of TPO (Chr2:1497591G>T NM_000547.5:c.1786G>T; p.Glu596*). Sanger sequencing validated the variant and confirmed segregation in Family 2 in autosomal-recessive manner. The variant is not listed in HGMD (public version) while 8 alleles (heterozygous) are reported in the ExAC database only in South Asian individuals (8181 individuals).
WES was performed for Family 3 using DNA from affected individual (IV:1) and his healthy parents (III-6 and III-7). All variants with minor allele frequency >1% were filtered out; we obtained 30 variants of which 10 were homozygous, 1 de novo, 13 compound heterozygous and 6 hemizygous. Analysis with different in silico prediction programs (MutationTaster, SIFT, PolyPhen2, PROVEAN) revealed 21 putatively pathogenic variants (6 homozygous, 1 de novo, 12 compound heterozygous and 2 hemizygous) in 14 different genes (Supplementary Table S1). For further analysis, we checked the expression of these genes in the brain (using UniGene (http://www.ncbi.nlm.nih.gov/unigene) and Human Protein Atlas (www.proteinatlas.org)), as well as the likely function of the gene, and finally we selected six variants from five different genes for further validation and segregation by Sanger sequencing. Only a homozygous missense variant in TPO, Chr2:1481273G>A; NM_003235.4: c.1235G>A; p.Arg412His, segregated with the phenotype in recessive manner. The variant was predicted to be pathogenic by four different in silico programs (PolyPhen2: possibly damaging; PROVEAN: damaging; SIFT: deleterious; and MutationTaster: disease causing), and the amino-acid residue is conserved across eukaryotic evolution, including insects and nematodes (Figure 3e). The variant is not listed in dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project,16 EVS (http://evs.gs.washington.edu/EVS/), ExAC or HGMD (public version; all accessed in December 2015).
Subsequently, free T3 (triiodothyronine) levels were tested in IV-1 and IV-3 from Family 3, using patient serum, by chemiluminescent technology with the following results: Patient IV-1 (Free T3, 4.9 pg ml−1); and Patient IV:3 (Free T3, 5.27 pg ml−1). The reference free T3 range for children aged 1–10 years is 0.92–2.48 ng ml−1 (Mayo Clinic, USA), thus hypothyroidism appears to be the likely cause of ID in these individuals. Unfortunately, we were unable to re-contact members from Family 1 and Family 2 and thus were unable to check thyroid function in these families.
Discussion
In this study, we investigated three families with ID. One family had a known mutation in TG while two families had novel mutations in TPO. Patients in Family 1 with the TG mutation had severe ID, goiter and prominent dysmorphic features. In Family 1, we used targeted sequencing, guided by the two HBD regions identified previously.8 Given the presence of goiter, TG was a good candidate gene and we identified a homozygous nonsense mutation that segregated with the phenotype. Two patients in Family 2 (II-1 and II-2) had moderate ID, and their dysmorphic features were less prominent. They did not have goiter. For Family 2, HBD information combined with WES identified a homozygous nonsense mutation in TPO, whereas for Family 3 WES was performed on proband and his unaffected parents and identified a homozygous missense mutation in TPO.
Dietary iodine is converted to iodide in the stomach before its absorption from the gastrointestinal tract to blood. It is transported from the blood stream across the follicular cells' basement membrane in the thyroid gland. This transport is facilitated by Na+/I− symporter—an intrinsic plasma membrane transporter protein.17 From the follicular cells, iodide is transported to the colloid in the lumen of the follicles. TG is a glycoprotein homodimer produced in thyrocytes that, after exocytosis into the lumen of the follicles, serves as a matrix for synthesis of thyroid hormones thyroxine (T4) and triiodothyronine (T3). TG is encoded by TG on chromosome 8, which is 270 Kb in length and has 48 exons.18 Only homozygous or compound heterozygous mutations are thought to lead to functional changes in TG gene that result in TDH (MIM 274700).
Iodide is oxidized to serve as iodizing agent. TPO catalyzes this step. TPO is also essential for incorporation of iodide onto tyrosine residues of TG and the coupling of iodotyrosines to generate T4 and T3. TPO is located on chromosome 2, spans about 150 Kb and consists of 17 exons and 16 introns.19
Normal TG and TPO functioning is essential for synthesis of T4 and T3.20 These hormones are essential in the normal development of brain.21 Mostly recessively inherited mutations in these genes cause functional changes in these hormones. For this reason, such mutations continue to be an important yet potentially preventable cause of ID in consanguineous populations. Recently, a homozygous loss copy number variation spanning several exons of TPO was identified in another Pakistani family ascertained with ID.22 Mutations in TG and TPO have been reported in the past for TDH (OMIM 188450 and 606765); however, in countries where health-care resources are limited and levels of consanguinity high, it is highly likely that this diagnosis would be frequently missed. Hence, TDH owing to mutation in either TG or TPO may be a relatively frequent cause of ID in such populations. Our data and previously reported data suggest that TDH is one the most common cause of ID in Pakistan.22 Families identified with segregating mutations in these genes should be counseled, and options for either genetic or blood-based screening for younger family members should be considered, and preventive therapies (for example, levothyroxine (L-thyroxine)) must be applied for those identified by genetic analysis as homozygotes or by blood screening as having hypothyroidism.
The prevalence of iodine deficiency as micronutrients in the general population is a problem in Pakistan. It can lead to hypothyroidism and developmental delay in infants and children. We are presenting genetic data that demonstrate that homozygous mutations can also contribute to thyroid disease and ID. In addition to public health campaigns to encourage use of iodized salt, physicians and health professionals should be aware of the possibility of genetic causes of hypothyroidism.
References
Loeber, J. G. Neonatal screening in Europe; the situation in 2004. J. Inherit. Metab. Dis. 30, 430–438 (2007).
Toublanc, J.-E. Comparison of epidemiological data on congenital hypothyroidism in Europe with those of other parts in the world. Horm. Res. 38, 230–235 (1992).
Bongers-Schokking, J. J., Koot, H. M., Wiersma, D., Verkerk, P. H. & de Muinck Keizer-Schrama, S. M. Influence of timing and dose of thyroid hormone replacement on development in infants with congenital hypothyroidism. J. Pediatr 136, 292–297 (2000).
Delange, F. Neonatal screening for congenital hypothyroidism results and perspectives. Horm. Res. 48, 51–61 (1997).
Cangul, H., Aycan, Z., Olivera-Nappa, A., Saglam, H., Schoenmakers, N. A., Boelaert, K. et al. Thyroid dyshormonogenesis is mainly caused by TPO mutations in consanguineous community. Clin. Endocrinol. 79, 275–281 (2013).
Park, S. M. & Chatterjee, V. K. K. Genetics of congenital hypothyroidism. J. Med. Genet. 42, 379–389 (2005).
Therrell, B. L., Padilla, C. D., Loeber, J. G., Kneisser, I., Saadallah, A., Borrajo, G. J. C. et al. Current status of newborn screening worldwide. Semin. Perinatol. 39, 171–187 (2015).
Rafiq, M. A., Ansar, M., Marshall, C. R., Noor, A., Shaheen, N., Mowjoodi, A. et al. Mapping of three novel loci for non-syndromic autosomal recessive mental retardation (NS-ARMR) in consanguineous families from Pakistan. Clin. Genet. 78, 478–483 (2010).
Lahiri, D. K. & Nurnberger, J. I. Jr . A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 19, 5444 (1991).
Lin, M., Wei, L.-J., Sellers, W. R., Lieberfarb, M., Wong, W. H. & Li, C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics 20, 1233–1240 (2004).
Seelow, D., Schuelke, M., Hildebrandt, F. & Nürnberg, P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res. 37, W593–W599 (2009).
Gazal, S., Sahbatou, M., Babron, M.-C., Génin, E. & Leutenegger, A.-L. FSuite: exploiting inbreeding in dense SNP chip and exome data. Bioinformatics 30, 1940–1941 (2014).
Mir, A., Sritharan, K., Mittal, K., Vasli, N., Araujo, C., Jamil, T. et al. Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum. Genet. 133, 975–984 (2014).
Rafiullah, R., Aslamkhan, M., Paramasivam, N., Thiel, C., Mustafa, G., Wiemann, S. et al. Homozygous missense mutation in the LMAN2L gene segregates with intellectual disability in a large consanguineous Pakistani family. J. Med. Genet. 53, 138–144 (2016).
Machiavelli, G. A., Caputo, M., Rivolta, C. M., Olcese, M. C., Gruñeiro-Papendieck, L., Chiesa, A. et al. Molecular analysis of congenital goitres with hypothyroidism caused by defective thyroglobulin synthesis. Identification of a novel c.7006C>T [p.R2317X] mutation and expression of minigenes containing nonsense mutations in exon 7. Clin. Endocrinol. (Oxf.) 72, 112–121 (2010).
Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., Handsaker, R. E. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Carrasco, N. Iodide transport in the thyroid gland. Biochim. Biophys. Acta 1154, 65–82 (1993).
Targovnik, H. M. in Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text, 10th edn (eds Braverman, L. E. & Cooper, D. S.) 74–92 (Lippincott Williams and Wilkins, Philadelphia, PA, USA, 2013).
Kopp, P. in Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text, 10th edn (eds Braverman, L. E. & Cooper, D. S.) 48–73 (Lippincott Williams and Wilkins, Philadelphia, PA, USA, 2013).
Vulsma, T. & De Vijlder, J. J. M. in Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text, 10th edn (eds Braverman, L. E. & Cooper, D. S.) 535–551 (Lippincott Williams and Wilkins, Philadelphia, PA, USA, 2013).
Van Vliet, G. & Deladoey, J. in Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text 10th edn (eds Braverman, L. E. & Cooper, D. S.) 787–789 (Lippincott Williams and Wilkins, Philadelphia, PA, USA, 2013).
Iqbal, Z., Neveling, K., Razzaq, A., Shahzad, M., Zahoor, M. Y., Qasim, M., et al. Targeted next generation sequencing reveals a novel intragenic deletion of the TPO gene in a family with intellectual disability. Arch. Med. Res 43, 312–316 (2012).
Acknowledgements
We thank the study participants and their families for their invaluable contributions to this study. This study was supported by a grant from the Canadian Institutes of Health Research to JBV (no. MOP-102758) and also by the Medical Faculty of the University Heidelberg. RR was supported by a scholarship from the German Academic Exchange Service (DAAD).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Mittal, K., Rafiq, M., Rafiullah, R. et al. Mutations in the genes for thyroglobulin and thyroid peroxidase cause thyroid dyshormonogenesis and autosomal-recessive intellectual disability. J Hum Genet 61, 867–872 (2016). https://doi.org/10.1038/jhg.2016.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.62
This article is cited by
-
Novel homozygous variant in the TPO gene associated with congenital hypothyroidism and mild-intellectual disability
Human Genome Variation (2020)
