
Abstract
Dravet syndrome (DS) is a severe childhood epilepsy typically caused by de novo dominant mutations in SCN1A. Although patients with DS frequently have neurocognitive abnormalities, the precise neural mechanisms responsible for their expression have not been elucidated. There are wide phenotypic differences among individuals with SCN1A mutations, suggesting that factors other than the SCN1A mutation modify the phenotype. Therefore, a well-controlled cellular model system is required to improve our understanding of the mechanisms underlying DS. Here we generated induced pluripotent stem cell (iPSC) lines from an individual with SCN1A mutation mosaicism, and separately cloned iPSC lines both with and without the SCN1A mutation. These clones theoretically have the same genetic backgrounds, except for the SCN1A gene, and should serve as an ideal pair for investigating the pathophysiology caused by SCN1A mutations. Quantitative reverse transcription-PCR and western blot analysis revealed higher tyrosine hydroxylase mRNA and protein expression levels in mutant neurons than in wild-type neurons. Moreover, dopamine concentrations in media collected from mutant neural cultures were higher than those from wild-type neural cultures. Our findings suggest that SCN1A mutation leads to changes in the dopamine system that may contribute to the behavioral abnormalities in DS.
Similar content being viewed by others

Introduction
Dravet syndrome (DS) is a childhood-onset intractable epilepsy mainly caused by de novo dominant mutations in SCN1A, encoding for the voltage-gated sodium channel NaV1.1. Patients with DS frequently have neurocognitive abnormalities including mental retardation, autistic behaviors, attention-deficit/hyperactive disorder and other psychiatric disorders.1 These abnormalities severely disrupt patients’ quality of life, so new treatments targeting them are needed. However, the precise neural mechanisms responsible for their expression in DS have not been elucidated.
Patient-derived induced pluripotent stem cell (iPSC) technologies have given us an opportunity to investigate the pathophysiology of human neural disorders in vitro throughout cellular developmental stages. However, as neurocognitive disorders generally have subtle phenotypes in vitro, it is difficult to distinguish disease-relevant phenotypic changes from background variation. In these cases, well-controlled cellular model systems are required. In this study, we generated iPSCs from an individual with SCN1A mutation mosaicism,2 and separately cloned iPSC lines both with and without the SCN1A mutation. These clones theoretically have the same genetic backgrounds, except for the SCN1A gene, and should serve as an ideal cellular model system for elucidating the genetic mechanisms of neurocognitive abnormalities caused by SCN1A mutation. Using this isogenic system, we aimed to determine whether the dopamine system is perturbed by SCN1A mutation.
Materials and Methods
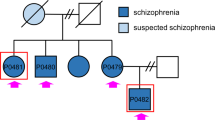
We previously described a family with two brothers who have DS.2 The two brothers (II-1 and II-2) had identical SCN1A truncation mutations, which were inherited from their mother (I-2), who is mosaic for the mutation (Figure 1a). In this study, we generated iPSCs from peripheral blood lymphocytes of the mother, who has experienced neither seizures nor neurocognitive disorders. Early clinical features of the brothers were described previously.2 The first son (II-1) had earlier seizure onset and more frequent seizures than the second son (II-2), and at 14 years old, speaks no meaningful words. He presents with autistic behaviors, hyperactivity and severe mental retardation. The second son is 11 years old now, and has demonstrated moderate mental retardation with attention-deficit/hyperactive disorder. Written informed consent was obtained from the donor, and ethical approval for this study was obtained from the Review Board of Kyoto Prefectural University of Medicine and Kyoto University. The lymphocytes were reprogrammed to iPSCs with the use of episomal vectors as previously described.3 Neuronal differentiation was performed by modifying previous procedures.4 Please see Supplementary Methods for more details. All primer sequences are shown in Supplementary Table S1.

Establishment of the isogenic cellular model system for Dravet syndrome. (a) Pedigree of the family. The first son (II-1) and the second son (II-2) are affected with Dravet syndrome. Their mother (I-2) exhibits SCN1A mutation mosaicism. (b) Mutation analysis for SCN1A. Mutant induced pluripotent stem cells (iPSCs) showed heterozygous mutation of SCN1A (c.[730G>T; 735G>T; 736A>T]), which resulted in a premature termination codon. (c) Phase contrast images of wild-type and mutant iPSCs growing on feeder cells. Scale bar, 500 μm. (d) Karyotype analysis of wild-type and mutant iPSC lines. (e) Quantitative PCR for the expression of pluripotent markers in iPSCs relative to human embryonic stem cells (KhES1). White columns show the copy numbers obtained with specific primers to the episomal vector of the indicated genes, black columns show the copy numbers obtained with primers for the coding sequence, which could detect both endogenous and episomal vector-derived transcripts. PCR with specific primers against the episomal vector of the indicated genes was not amplified in these iPSC lines, proving the disappearance of episomal vectors. Vector only was used as positive control. (f) Teratoma formation shows tissues from three germ layers. (g) SCN1A mRNA expression levels of wild-type and mutant iPSCs. Values are mean±s.d. *P=0.015 (n=3 independent cultures).
Results and Discussion
We isolated individual iPSC clones, expanded them and performed a mutation analysis on SCN1A. Two of 11 isolated clones had a heterozygous mutation of the SCN1A gene, whereas the rest of the clones were wild type (Figure 1b). We selected one SCN1A mutant and one wild-type clone for subsequent analyses. Both clones showed characteristic human embryonic stem cell-like morphology (Figure 1c), normal karyotype (Figure 1d) and expression of endogenous pluripotent markers similar to control embryonic stem cell (Figure 1e). Expression of introduced transgenes was not detected (Figure 1e). The pluripotency of both iPSC clones was confirmed by the presence of cell derivatives of all three germ layers in a teratoma formation assay (Figure 1f). Quantitative reverse transcription (RT)-PCR analysis revealed that SCN1A mRNA expression levels of mutant iPSCs were ~50% lower than that of wild-type iPSCs (Figure 1g). Cells differentiated in our culture condition were regionally specified as telencephalic, diencephalic and midbrain fates (Figure 2a and b). Both mutant and wild-type differentiated cultures were able to produce mature-looking neurons expressing the neuronal marker βIII-tubulin (Figure 2c). Quantitative RT-PCR and western blot analysis revealed that both TUBB3 mRNA and βIII-tubulin protein expression levels were similar between mutant and wild-type neural aggregates on day 21 (Figures 2d and e). Together with the further RT-PCR analysis of neuronal markers evaluating maturation state and cell types (Figure 2f), our data suggest that there was no prominent difference in the neuronal differentiation propensity between mutant and wild-type cells. We used this model to determine whether the SCN1A mutation leads to perturbation in dopaminergic activity. Quantitative RT-PCR and western blot analysis of mutant neural aggregates on day 21 revealed that both tyrosine hydroxylase (TH) mRNA and TH protein expression levels were higher than those of wild-type neural aggregates (Figures 3a and b). We measured dopamine in the media collected from the neural cultures and found that the mutant neurons secreted about 2.5 times more dopamine than wild-type neurons (Figure 3c). Next, to determine whether the increased dopamine production in mutant neurons is caused by a change in the composition of the neuronal population, we counted the number of TH-positive neurons in the cultures. We found that 4.97% (47/946 cells) of the wild-type neurons and 5.24% (51/974 cells) of the mutant neurons expressed TH, indicating that the proportion of TH-positive neurons was not different between mutant and wild type (Figures 3d and e).

Similar potency of neuronal differentiation in wild-type and mutant induced pluripotent stem cells (iPSCs). (a) Schematic diagram of neuronal differentiation and representative images during neuronal differentiation: iPSC (left), neural aggregates on day 21 (middle) and dissociated neurons (right). Day numbers indicate the days of differentiation after neural induction. Scale bar, 100 μm. (b) Reverse transcription (RT)-PCR analysis for the expression of regional markers in central nervous system. Neural aggregates on day 21 expressed telencephalic (TBR1, FOXG1, OTX2 and NKX2.1), diencephalic (SIX3) and midbrain (LMX1A) markers, but not hindbrain (KROX20) or spinal cord (HOXB4) markers. (c) Both wild-type and mutant iPSC-derived neurons expressed the neuronal marker βIII-tubulin (green). Scale bar, 200 μm. (d) Quantitative RT-PCR analysis of TUBB3 mRNA in neural aggregates on day 21. Values are mean±s.d. (n=6 independent cultures). (e) Western blot analysis of βIII-tubulin protein in neural aggregates on day 21. Values are mean±s.d. (n=3 independent cultures). (f) Quantitative RT-PCR analysis of different cell identity markers in neural aggregates on day 21. The expression of neuronal (MAP2, SYN1, VGLUT1 and VGAT), cortical layer VI (TBR1) and layer II–IV (SATB2) markers is shown. Values are mean±s.d. (n=6 independent cultures).

Aberrant dopamine levels in SCN1A mutant neurons. (a) Quantitative reverse transcription-PCR analysis of tyrosine hydroxylase (TH) mRNA. TH mRNA expression levels of neural aggregates on day 21 were significantly higher in mutant compared with wild type. Values are mean±s.d. *P=0.019 (n=6 independent cultures). (b) Western blot analysis of TH protein. TH protein expression levels of neural aggregates on day 21 were significantly higher in mutant compared with wild type. Values are mean±s.d. *P=0.045 (n=3 independent cultures). (c) Dopamine concentration in the media collected from the neural cultures. Mutant neurons secreted more dopamine than wild-type neurons. Values are mean±s.d. **P=0.00392 (n=4 independent cultures). (d) Representative images of neurons stained with antibodies specific to TH and βIII-tubulin. TH-positive neurons (TH+/βIII-tubulin+) are indicated by arrows and TH-negative neurons (TH−/βIII-tubulin+) are indicated by arrowheads. Scale bar, 50 μm. (e) The percentage of TH-positive neurons (TH+/βIII-tubulin+) in mutant and wild-type neuronal cultures (51/974 and 47/946 neurons, respectively, in three independent cultures).
Patient-derived iPSCs have been used in several previous studies for DS.5, 6, 7 However, these studies compared patient-derived cells with normal cells of other individuals, and focused on the mechanisms of seizure susceptibility rather than the neurocognitive abnormalities. Our isogenic cellular model system, which allows ‘clean’ detection of SCN1A mutation-caused effects, is well-suited for detecting the subtle cellular phenotypic changes causing neurocognitive disorders. Previous studies have shown that mice in which Scn1a was selectively knocked down in the basal forebrain via RNA interference had perturbed learning and memory without seizures.8 In addition, mice with Scn1a haploinsufficiency demonstrated impaired GABAergic neurotransmission, autistic-like behaviors and phenotypic rescue, with low doses of clonazepam, a positive allosteric modulator of GABAA receptors.9 These studies indicate that SCN1A mutations cause neurocognitive dysfunctions through a mechanism independent of seizures, and impaired GABAergic neurotransmission is crucial for the neurocognitive dysfunction in DS. However, different pathological mechanisms might underlie neurocognitive dysfunctions in SCN1A-related epilepsies. Recently, application of human iPSC-derived neurons to the in vitro assessment of neurotransmitter secretion related to neuropsychiatric disorders was reported.10 In this study, we found increased basal secretion of dopamine in mutant iPSC-derived neurons. The dopaminergic system is strongly involved in the pathogenesis of autism.11 Indeed, various dopamine-receptor antagonists are effective for behavioral problems in autistic children.12 Although it is likely that autistic features observed in DS patients have a multifactorial etiology, our findings seem suggestive of a possible contribution of the dopaminergic system to their expression. Intriguingly, similar to our DS model, iPSC-derived neuron from individuals with Timothy syndrome, a neurodevelopmental disorder with autistic features caused by the l-type calcium channel gene mutation, showed elevated expression of TH and increased production of dopamine.13 However, in contrast to Timothy syndrome neurons, the number of TH-positive dopaminergic neurons in the differentiated culture revealed no difference between mutant and wild type. The precise reason for this discrepancy is unknown, but methodological differences during culture, such as differences in the protocol of neuronal differentiation and the timing of the analysis, might contribute to this phenotypic difference.
There are several obvious limitations of this study. The first one is that our findings relate to a single case and should be confirmed in additional individuals. However, individuals with SCN1A mutation mosaicism are too rare to enroll additional cases in the study. Instead, genome-editing technologies can be used to generate additional isogenic cell lines.14 The second one is that the underlying mechanism of elevated TH expression in SCN1A mutant neurons remains obscure. The TH promoter contains numerous transcription factor-binding sites that permit mRNA levels to be regulated by various stimulations.15 Among them cAMP response element-binding protein (CREB), which is activated through the phosphorylation at its regulatory site on serine residue 133, is one of the most important factors for the control of TH expression.15 To determine whether the increase in TH expression was a result of CREB activation, we performed western blot analysis using an antibody that recognizes p-CREB. However, the level of p-CREB was not elevated in mutant neurons (data not shown), indicating that other factors might lead to changes in TH expression. Finally, our findings were the opposite of those from the study on rats with a N1417H–Scn1a mutation, where dopamine levels were reduced in the reward pathway.16 However, N1417H missense mutation, which arose from random mutagenesis, does not represent SCN1A mutations found in human patients. Furthermore, it should be noted that there are several differences in the regulation of TH expression between rodents and humans.15 Indeed, discrepancy in TH expression between mice and humans was also observed in Timothy syndrome models.13 Although further studies are required to resolve such limitations, this study provided additional evidence linking aberrant dopaminergic activity to ion channel gene defects. In conclusion, our isogenic system can contribute to the understanding of the pathophysiology of neurocognitive abnormalities in DS and to the development of more effective therapies in the future.
References
Wolf, M., Casse-Perrot, C. & Dravet, C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 47 (Suppl 2), 45–48 (2011).
Morimoto, M., Mazaki, E., Nishimura, A., Chiyonobu, T., Sawai, Y., Murakami, A. et al. SCN1A mutation mosaicism in a family with severe myoclonic epilepsy in infancy. Epilepsia 47, 1732–1736 (2006).
Okita, K., Matsumura, Y., Sato, Y., Okada, A., Morizane, A., Okamoto, S. et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412 (2011).
Morizane, A., Doi, D., Kikuchi, T., Nishimura, K. & Takahashi, J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J. Neurosci. Res. 89, 117–126 (2011).
Jiao, J., Yang, Y., Shi, Y., Chen, J., Gao, R., Fan, Y. et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum. Mol. Genet. 22, 4241–4252 (2013).
Liu, Y., Lopez-Santiago, L. F., Yuan, Y., Jones, J. M., Zhang, H., O’Mally, H. A. et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol. 74, 128–139 (2013).
Higurashi, N., Uchida, T., Lossin, C., Misumi, Y., Okada, Y., Akamatsu, W. et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol. Brain 6, 19 (2013).
Bender, A. C., Natola, H., Ndong, C., Holmes, G. L., Scott, R. C. & Lenck-Santini, P. P. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol. Dis. 54, 297–307 (2013).
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B. et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390 (2012).
Hook, V., Brennand, K. J., Kim, Y., Toneff, T., Funkelstein, L., Lee, K. C. et al. Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Reports 3, 531–538 (2014).
Nguyen, M., Roth, A., Kyzar, E. J., Poudel, M. K., Wong, K., Stewart, A. M. et al. Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD). Neurochem. Int. 66, 15–26 (2014).
McCracken, J. T., McGough, J., Shah, B., Cronin, P., Hong, D., Aman, M. G. et al. Risperidone in children with autism and serious behavioral problems. N. Engl. J. Med. 347, 314–321 (2002).
Pasca, S. P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Pasca, A. M. et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662 (2011).
Chen, W., Liu, J., Zhang, L., Xu, H., Guo, X., Deng, S. et al. Generation of the SCN1A epilepsy mutation in hiPS cells using the TALEN technique. Sci. Rep. 4, 5404 (2014).
Tekin, I., Roskoski, R. Jr, Carkaci-Salli, N. & Vrana, K. E. Complex molecular regulation of tyrosine hydroxylase. J. Neural Transm. 1221, 1451–1481 (2014).
Ohmori, I., Kawakami, N., Liu, S., Wang, H., Miyazaki, I., Asanuma, M. et al. Methylphenidate improves learning impairments and hyperthermia-induced seizures caused by an Scn1a mutation. Epilepsia 55, 1558–1567 (2014).
Acknowledgements
We are grateful to the family for their cooperation in this study. This study was supported by Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI Grant No. 15K09628) and the Program for Intractable Diseases Research utilizing Disease-specific iPS cells of the Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Maeda, H., Chiyonobu, T., Yoshida, M. et al. Establishment of isogenic iPSCs from an individual with SCN1A mutation mosaicism as a model for investigating neurocognitive impairment in Dravet syndrome. J Hum Genet 61, 565–569 (2016). https://doi.org/10.1038/jhg.2016.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.5
This article is cited by
-
From molecules to medicines: the dawn of targeted therapies for genetic epilepsies
Nature Reviews Neurology (2018)
