
Abstract
Pontocerebellar hypoplasia (PCH) is characterized by hypoplasia and atrophy of the cerebellum, variable pontine atrophy, microcephaly, severe mental and motor impairments and seizures. Mutations in 11 genes have been reported in 8 out of 10 forms of PCH. Recessive mutations in the mitochondrial arginyl-transfer RNA synthetase gene (RARS2) have been recently associated with PCH type 6, which is characterized by early-onset encephalopathy with signs of oxidative phosphorylation defect. Here we describe the clinical presentation, neuroimaging findings and molecular characterizations of two siblings with a clinical diagnosis of PCH who displayed a novel variant (c.-2A>G) in the 5′-UTR of the RARS2 gene in the homozygous state. This variant was identified through next-generation sequencing testing of a panel of nine genes known to be involved in PCH. Gene expression and functional studies demonstrated that the c.-2A>G sequence change directly leads to a reduced RARS2 messenger RNA expression in the patients by decreasing RARS2 promoter activity, thus providing evidence that mutations in the RARS2 promoter are likely to represent a new causal mechanism of PCH6.
Similar content being viewed by others


Introduction
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of autosomal recessive neurodegenerative disorders characterized by an abnormally small cerebellum and brainstem, severe motor impairments and intellectual disability.1 To date, 10 subtypes have been described (PCH1–10). Clinical features and the severity of the volume reduction of the cerebellar hemispheres, the vermis and of the pons, vary between subtypes. Eleven genes have been reported causative in 8 out of the 10 forms of PCH with the exception of PCH5 and PCH7. Mutations in VRK1 and EXOSC3 have been reported in patients with PCH type 1, which is characterized by central and peripheral motor dysfunction associated with anterior horn cell degeneration resembling infantile spinal muscular atrophy.2, 3, 4 Mutations in the tRNA-splicing endonuclease (TSEN) complex (TSEN54, TSEN2, TSEN34) and SEPSECS have been described in PCH type 2,5, 6, 7 which presents with progressive microcephaly from birth combined with extrapyramidal dyskinesias.8 Mutations of TSEN54 have also been identified in patients with PCH type 4,6 which is characterized by hypertonia, joint contractures, olivopontocerebellar hypoplasia and early death.6 Most recently, a novel hemizygous frameshift mutation in the CASK gene was identified in a male patient with PCH3, early myoclonic epilepsy and tetralogy of Fallot.9 In 2007, Edvardson et al. first reported a homozygous intronic mutation in the RNA synthetase gene (RARS2) in three patients from a consanguineous family with Sephardic Jewish ancestry, who presented with severe infantile encephalopathy associated with PCH and multiple mitochondrial respiratory-chain defects (PCH6).10 Within the past 2 years, PCH8, PCH9 and PCH10 have been described and the associated genes CHMP1A, AMPD2 and CLP1 have also been identified.11, 12, 13, 14
Among all forms of PCH, only patients with PCH6 present with progressive atrophy of the supra and subtentorial brain structures and white matter depletion. Patients with PCH6 may also show multiple generalized reductions in the respiratory-chain enzyme activities in muscle and elevated blood and cerebrospinal fluid lactate levels.15 To date, more than 10 patients with PCH6 have been reported with mutations in RARS2, which encodes a mitochondrial arginyl-tRNA synthetase, an enzyme essential for all proteins synthesized in mitochondria.15, 16, 17, 18
The promoter is a regulatory region of the DNA. It is located upstream of genes and has an important role in transcriptional regulation. Mutations occurring in the promoter region of a gene may disrupt the normal processes of gene activation by disturbing the ordered recruitment of transcription factors at the promoter. An estimated 1% of single base-pair substitutions that occur within gene promoter regions cause human genetic disease.19 Examples of disorders that can be associated with promoter mutations include β-thalassemia, pyruvate kinase deficiency, familial hypercholesterolemia and hemophilia.20 However, no mutations have been reported in the promoter of RARS2 and other PCH-associated genes. In this study, we investigated a cohort of 48 patients clinically diagnosed with PCH and evaluated them for mutations in nine genes (CASK, CHMP1A, EXOSC3, RARS2, SEPSECS, TSEN2, TSEN34, TSEN54 and VRK1) known to be associated with PCH. Disease-causing mutations were identified in nine patients. Most importantly, we present the first reported mutation (c.-2A>G) within the promoter sequence of RARS2 in two siblings with PCH6, and also demonstrate that this variant is associated with a reduced RARS2 expression.
Materials and methods
DNA isolation
Peripheral blood samples from patients were referred to molecular diagnostic laboratory at the University of Chicago for PCH gene panel analysis. Genomic DNA was isolated from blood leukocytes on the AutoGenFlex STAR robotic workstation (Autogen, Holliston, MA, USA) or using the MagNA Pure Compact DNA isolation system (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instructions.
Next generation sequencing
Genomic coordinates were identified for all target regions in 158 genes related to a collection of neurological conditions, including nine genes reported to be causative of PCH (CASK, CHMP1A, EXOSC3, RARS2, SEPSECS, TSEN2, TSEN34, TSEN54 and VRK1). Target regions included the coding sequence plus 50 bp flanking intronic or UTR sequence. This design was submitted to RainDance Technologies for primer synthesis and emulsification. Three micrograms of patient DNA was sheared to 5 kb fragments (Covaris S2, Woburn, MA, USA). PCR master mix was created, emulsified, merged with the primer-pair droplets and dispensed as an emulsion into a PCR tube, using the RDT1000 (RainDance Technologies, Lexington, MA, USA). Samples were PCR amplified, emulsion was destabilized and PCR products were purified using a MinElute column (Qiagen, Frederick, MD, USA). Pooled amplicons were end-repaired (NEBNext End Repair Module), randomly concatenated to 3 kb fragments (NEBNext Quick Ligation Module), and sheared to ~300 bp. DNA fragments were built into sequence-ready libraries using the Illumina TruSeq protocol, and 2 × 150 paired-end reads were generated on an Illumina MiSeq. Fastq files were aligned against the hg18 reference sequence using BWA, yielding a minimum of 200 × average depth across the targeted regions. Variants were called using SAMTOOLS and GATK, and filtered to focus analysis on those variants within the PCH genes.
RNA extraction and quantitative real-time PCR analysis
Total RNA was isolated from peripheral blood samples using the miRNeasy kit (Qiagen). For messenger RNA (mRNA) expression, 150 ng of RNA was reverse-transcribed into complementary DNA in a total reaction volume of 10 μl using the Qiagen’s RT kit according to the manufacturer’s instructions. Quantitative real-time PCR analysis was performed using 0.5 μl of complementary DNA using SYBR green PCR. Actin, beta (ACTB) was used as an endogenous control. All of the samples were run in triplicate.
Plasmid construction, transient transfection and promoter luciferase assay
The proximal promoter (a total of 672 bp including exon 1 and 198 bp downstream and 378 bp of upstream of exon 1) of the human RARS2 gene containing wild-type (pGL4-WT) or the mutant (c.-2A>G, pGL4 –Mut) 5′-UTR sequences were cloned into a pGL4 basic vector. For transfection, HEK293T cells were plated in 96-well plates at a concentration of 6000 cells per well in triplicate for each condition. After overnight incubation, cells were transfected with 20 ng of the pGL4-WT, pGL4-Mut or pGL4 empty vector, and co-transfected with 1 ng of pMIR-REPORT Beta-galactosidase Reporter Control Vector (Ambion, Austin, TX, USA) using Effectene Transfection Reagent (Qiagen) according to the manufacturer’s protocol. Cells were lysed and firefly luciferase and β-galactosidase activities were detected using Dual-Light Combined Reporter Gene Assay System (Applied Biosystems, Foster City, CA, USA) 48 h post transfection. Firefly luciferase activity was normalized to β-galactosidase activity for each transfected well. Each experiment was performed in triplicate and repeated three times.
Magnetic resonance imaging (MRI) analysis
Postnatal MRI studies were obtained on either a 1.5 or a 3.0 T scanner (GE Healthcare, Waukesha, WI, USA); the following sequences were acquired: sagittal 3D volume T1-weighted images, axial and coronal T2-weighted images, axial proton density (or FLAIR). Prenatal scans were obtained on a 1.5 T scanner (GE Healthcare); coronal, sagittal and axial T2-weighted image sequences were acquired.
Results
Sequence results
Sequence analysis of a cohort of 48 patients with various PCH forms identified pathogenic or likely pathogenic sequence changes in nine patients (Table 1). Mutations in TSEN54 were identified in six patients. The previously reported c.919G>T (p.Ala307Ser) founder mutation in TSEN546 was observed in all six patients in the homozygous or compound heterozygote state. Two unrelated patients (Pt2 and Pt5) harbored a CASK c.82C>T (p.Arg28*) truncating mutation, which has not been reported previously. Two EXOSC3 mutations (c.112del (p.Glu38Asnfs*16) and c.238G>T (p.Val80Phe)) were observed in Pt7, and were confirmed to be in compound heterozygous state by parental testing. Interestingly, a single nucleotide change was identified in the 5′-UTR of RARS2 (c.-2A>G) in Pt10 in the homozygous state as well as in this patient’s similarly affected sibling (Table 1, Figures 1a and b). This sequence change has not been observed in unaffected individuals (Exome Sequencing Project, 1000 Genome Project), or in other patients with RARS2- related disorders.

The c.-2A>G sequence change in RARS2 identified in the patients. (a) The sequence change identified by NGS in proband Pt10. (b) Sanger sequencing confirmation showing the sequence change in homozygous state in Pt10 and his sister Pt11, and in heterozygous state in their parents. A full color version of this figure is available at the Journal of Human Genetics journal online.
Clinical findings
Proband (Pt10)
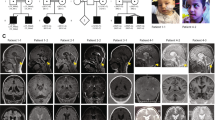
The proband is the first child of Hispanic parents and was delivered at full term following a pregnancy complicated by maternal bleeding at 2–3 months gestation and maternal hypertension. Physical examination at birth was normal. He was reported to have normal development up to ~6 months of age. At 9 months of age he presented with hypotonia, seizures, loss of milestones and clonus. MRI findings at 9 months were consistent with PCH, demonstrating small posterior fossa, vermis hypoplasia, small pons and atrophy of cerebellar hemispheres, as well as prominent subarachnoid space overlying frontotemporal convexities (Figures 2a–c). At 3 years of age, clinical molecular testing for PCH was preformed and a novel homozygous sequence change, c.-2A>G in the 5′-UTR of RARS2 was identified (Figures 1a and b). At 4.5 years the patient is unable to sit, has microcephaly and continues to have poor head control. Head circumference continues to be at the 50th percentile. He has had progressive vision loss, no functional speech and refractory partial epilepsy, which has been difficult to control. Further clinical information is provided in Table 2. Repeat brain MRIs at 27 and 50 months have not shown significant progression (not shown).

Pt10’s MRI obtained at 9 months. Coronal T2-weighted image through the cerebellar hemispheres (a), sagittal T1-weighted image through midline (b) and sagittal T1-weighted image off midline (c) images demonstrate severe atrophy of vermis (superior aspect more affected than inferior aspect) and moderate-to-severe atrophy of the cerebellar hemispheres. There is moderate atrophy of the dorsal pons. Fetal MRI at 22 weeks fetal age of Pt11 (Pt10’s sibling): coronal T2-weighted image (d) and sagittal T2-weighted image (transverse measurement 22 mm) (e) images demonstrate normal appearing cerebellum (height is 10 mm) with normals measuring 22+2 and 10 +1,27 respectively. MRI obtained in Pt 11 at 7 months: coronal T2-weighted image through the cerebellar hemispheres (f), sagittal T1-weighted images through midline (g) and sagittal T2-weighted image off midline (h) images demonstrate severe atrophy of the cerebellar vermis (superior aspect more affected than inferior aspect); moderate atrophy of the cerebellar hemispheres; and mild atrophy of the dorsal pons.
Proband’s sibling (Pt11)
Patient 11 (Pt11), the second child of the parents, is a girl born at full term by Cesarean section owing to fetal distress. Prenatal fetal MRI demonstrated no brain abnormalities in utero (Figures 2d and e). She did not gain milestones in infancy and was noted to have hypotonia at 6 months. Brain imaging with MRI at 6 months showed cerebellar hypoplasia (Figures 2f–h). Mutation analysis of RARS2 showed the same c.-2A>G sequence change in the homozygous state as her sibling (Figure 1b). Her head circumference was at the 10th percentile at 18 months of age. At 10 months of age she was able to sit with support and had started to babble. She had abnormal abduction of her right eye, no amblyopia and normal vision, and no history of seizures. Additional clinical information is provided in Table 2.
The patients’ parents are not known to be related but are both carriers of the RARS2 c.-2A>G sequence change (Figure 1b). Except for these two siblings, no other family members in three generations are reported to have developmental delay, intellectual disability, cerebral palsy or early death.
Gene expression and promoter functional studies
We investigated the possibility that the c.-2A>G variant in the 5′-UTR of the RARS2 gene may be located within the promoter region. The 5′-UTR of RARS2 is embedded in a transcription factor binding site as shown in the UCSC genome browser (Figure 3a), indicating that it maybe in a promoter region. We also found that the 5′-UTR is included in the promoter of RARS2 through searching the Eukaryotic Promoter Database (EPDnew), which is a series of species-specific databases of experimentally validated promoters. These evidences raised the possibility that a sequence change in the 5′-UTR of RARS2 might affect gene expression. To address this question, we performed quantitative real-time PCR analysis to detect the mRNA expression level of RARS2 in the peripheral blood of the two siblings. As shown in Figure 3b, the mRNA expression level of RARS2 in both Pt10 and Pt11 was significantly downregulated compared with the normal controls (P<0.05). There is no significant difference in the RARS2 expression level between the siblings. This suggested that the c.-2A>G sequence change may lead to reduced mRNA expression of RARS2 in these patients.

(a) The relative position of c.-2A>G sequence change in RARS2. (b) qRT-PCR analysis of the mRNA expression of RARS2 in Pt10, Pt11 and two normal controls. A full color version of this figure is available at the Journal of Human Genetics journal online.
To further clarify the effects of the RARS2 c.-2A>G sequence change, we conducted a promoter luciferase assay. We cloned the wild-type (pGL4-WT) and the c.-2A>G mutant (pGL4 –Mut) 5′-UTR sequences upstream of a reporter luciferase gene in a pGL4 basic promoterless luciferase reporter vector. The constructs were transiently transfected into HEK293T cells, which were derived from human embryonic kidney cells. Luciferase activity was detected 48 h after transfection. Transfection with pGL4–Mut resulted in 40% reduction of promoter activity compared with that of pGL4-WT (Figure 4). This demonstrated that the c.-2A>G sequence change directly reduced the RARS2 promoter activity.

Promoter luciferase assay of wild-type and mutant alleles of RARS2. pGL4, empty vector; pGL4-WT, a fragment of wild-type allele of RARS2 cloned into a pGL4 vector; and pGL4 –Mut, the c.-2A>G mutant allele of RARS2 cloned into a pGL4 vector. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
We examined a cohort of 48 individuals with PCH and identified disease-causing mutations in nine patients. Six of these patients had mutations in the TSEN54 gene, and the common founder mutation in this gene, c.919G>T (p.Ala307Ser)6 was identified in 11 of 12 (92%) alleles. These results further demonstrate that TSEN54 is the most common gene involved in PCH development.
Interestingly, we identified a novel single nucleotide transition, (A→G) in the RARS2 gene at position c.-2 in a patient and his sibling affected with PCH. A genetic diagnosis was supported by the demonstration that this sequence change segregated with the disease in the family and the sequence change was absent in a large group of controls. Our gene expression assays demonstrated that the mutation leads to a 40% reduction of RARS2 mRNA expression.
The RARS2 gene encodes the mitochondrial arginine-tRNA synthetase (Edvardson, S. 2007) that is essential for all proteins synthesized in mitochondria.15, 16, 17, 18 To date, all previously reported RARS2 mutations affect coding regions or splice sites. These mutations cause reduced RARS2 expression or low enzyme activity.10, 15 Cassandrini et al. reported three families with RARS2 missense mutations. In those families, the mRNA expression of mutated RARS2 mutations was ~60–70% of the normal controls when measured by qPCR in cultured skin fibroblasts. Functional complementation studies of those mutations in Saccharomyces cerevisiae showed that the c.1406G>A (p.Arg469His) abolished respiration, whereas the c.734G>A (p.Arg245Gln) displayed a reduced growth on non-fermentable YPG medium.15 This suggests that the observation of 40% reduction of RARS2 expression in our study likely results in a reduced mitochondrial arginyl-tRNA synthetase enzyme activity.
The 5′-UTR of RARS2 is embedded in a CpG island, which is a site of transcription initiation for most genes21 (Figure 3a). The 5′-UTR of RARS2 is also enriched with acetylation of histone H3, lysine 27 (H3K27Ac), a histone modification mark that is highly correlated with open chromatin and gene transcription22, 23 (Figure 3a). These observations support that the 5′-UTR of RARS2 is located in the promoter region. In fact, the 5′-UTR of RARS2 is reported within the promoter of RARS2 by the promoter database EPDnew. The promoter region of a gene contains binding sites for general transcription factors and RNA polymerase II. Single base-pair substitutions in the promoter region have been shown to disrupt the normal processes of gene activation and transcriptional initiation and lead to dysregulation of mRNA and protein expression. In our promoter luciferase assay, we showed that the c.-2A>G sequence change in the 5′-UTR of RARS2 significantly reduced the RARS2 promoter activity, which led to decreased RARS2 mRNA expression.
In addition, the c.-2A>G change in RARS2 is located within the Kozak sequence, which is involved in the initiation of translation and determines the translation efficiency (Kozak,1987a, 1987b). In humans, the consensus is GCCGCC(A/G)(C/A)CAUGGCG, with the start codon being essential and other positions within the Kozak sequence acting to enhance the affinity for the ribosome, such as G +4 and a purine on −3.24, 25 A spectrum of diseases, including autoimmune disease and cancer have been associated with Kozak sequence mutations.26 It is very likely that a mutation at the c.2 position of RARS2 can impair translation efficiency and lead to further reduced expression of RARS2 protein level.
It is also noted that the clinical features observed in this two siblings appear to be less severe than those from the previously reported patients with missense mutations in RARS2 who presented with severe hypotonia at birth or within a few days of life.10, 15, 16 The two siblings reported in this study showed normal development until 6 months of age. Blood and cerebrospinal fluid lactate levels in the siblings were not elevated as shown in other patients.10, 15, 16 It is possible that there is a higher level of residual expression of RARS2 in patients with promoter mutations than those with missense mutations. The expression of RARS2 level did not show significant difference between the two siblings, even though the clinical features of Pt11 appear to be milder than Pt10. The phenotypic variability within the affected siblings in this family may also indicate a role for environment, stochastic events, genetic background or a combination of these influences.
Taken together, the clinical features of the patients, the results of genetic testing and functional studies, we believe it is plausible that the c.-2A>G substitution in the RARS2 sequence has a pathogenic role in PCH for the following reasons: (1) the mutation segregates with PCH in the family; (2) the mutation has not been observed in unaffected individuals (Exome Sequencing Project, 1000 Genome Project); (3) the mutation occurs at a highly conserved position within the promoter sequence; (4) the mutation impairs RARS2 expression; and (5) the effect of other known mutations in RARS2 appear to affect RARS2 expression and mitochondrial arginyl-tRNA synthetase activity, which is similar to that of the mutation in this family.
This is the first study that has shown a single nucleotide change within the promoter of RARS2 reduces RARS2 expression by decreasing promoter activity in PCH patients. This mutation interferes with the mechanisms controlling the expression of RARS2 and affects mitochondrial arginyl-tRNA synthetase activity, thus providing insights for a new causal mechanism of PCH6 and expanding the RARS2 mutational spectrum.
References
Dyment, D. A., Sawyer, S. L., Chardon, J. W. & Boycott, K. M. Recent advances in the genetic etiology of brain malformations. Curr. Neurol. Neurosci. Rep. 13, 364 (2013).
Renbaum, P., Kellerman, E., Jaron, R., Geiger, D., Segel, R. & Lee, M. et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am. J. Hum. Genet. 85, 281–289 (2009).
Wan, J., Yourshaw, M., Mamsa, H., Rudnik-Schoneborn, S., Menezes, M. P. & Hong, J. E. et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 44, 704–708 (2012).
Barth, P. G. Pontocerebellar hypoplasia—how many types? Eur. J. Paediatr. Neurol. 4, 161–162 (2000).
Agamy, O., Ben Zeev, B., Lev, D., Marcus, B., Fine, D. & Su, D. et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 87, 538–544 (2010).
Budde, B. S., Namavar, Y., Barth, P. G., Poll-The, B. T., Nurnberg, G. & Becker, C. et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 40, 1113–1118 (2008).
Ben-Zeev, B., Hoffman, C., Lev, D., Watemberg, N., Malinger, G. & Brand, N. et al. Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J. Med. Genet. 40, e96 (2003).
Barth, P. G., Blennow, G., Lenard, H. G., Begeer, J. H., van der Kley, J. M. & Hanefeld, F. et al. The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly, and extrapyramidal dyskinesia (pontocerebellar hypoplasia type 2): compiled data from 10 pedigrees. Neurology 45, 311–317 (1995).
Nakamura, K., Nishiyama, K., Kodera, H., Nakashima, M., Tsurusaki, Y. & Miyake, N. et al. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 36, 272–273 (2014).
Edvardson, S., Shaag, A., Kolesnikova, O., Gomori, J. M., Tarassov, I. & Einbinder, T. et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81, 857–862 (2007).
Mochida, G. H., Ganesh, V. S., de Michelena, M. I., Dias, H., Atabay, K. D. & Kathrein, K. L. et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat. Genet. 44, 1260–1264 (2012).
Akizu, N., Cantagrel, V., Schroth, J., Cai, N., Vaux, K. & McCloskey, D. et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 154, 505–517 (2013).
Schaffer, A. E., Eggens, V. R., Caglayan, A. O., Reuter, M. S., Scott, E. & Coufal, N. G. et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663 (2014).
Karaca, E., Weitzer, S., Pehlivan, D., Shiraishi, H., Gogakos, T. & Hanada, T. et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157, 636–650 (2014).
Cassandrini, D., Cilio, M. R., Bianchi, M., Doimo, M., Balestri, M. & Tessa, A. et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J. Inherit. Metab. Dis. 36, 43–53 (2013).
Rankin, J., Brown, R., Dobyns, W. B., Harington, J., Patel, J. & Quinn, M. et al. Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am. J. Med. Genet. A 152A, 2079–2084 (2010).
Namavar, Y., Barth, P. G., Kasher, P. R., van Ruissen, F., Brockmann, K. & Bernert, G. et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134, 143–156 (2011).
Glamuzina, E., Brown, R., Hogarth, K., Saunders, D., Russell-Eggitt, I. & Pitt, M. et al. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J. Inherit. Metab. Dis. 35, 459–467 (2012).
Cooper, D. N. Human gene mutation in pathology and evolution. J. Inherit. Metab. Dis. 25, 157–182 (2002).
de Vooght, K. M., van Wijk, R. & van Solinge, W. W. Management of gene promoter mutations in molecular diagnostics. Clin. Chem. 55, 698–708 (2009).
Deaton, A. M. & Bird, A. CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011).
Tie, F., Banerjee, R., Stratton, C. A., Prasad-Sinha, J., Stepanik, V. & Zlobin, A. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 (2009).
Pasini, D., Malatesta, M., Jung, H. R., Walfridsson, J., Willer, A. & Olsson, L. et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38, 4958–4969 (2010).
Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148 (1987).
Nakagawa, S., Niimura, Y., Gojobori, T., Tanaka, H. & Miura, K. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 36, 861–871 (2008).
Wolf, A., Caliebe, A., Thomas, N. S., Ball, E. V., Mort, M. & Stenson, P. D. et al. Single base-pair substitutions at the translation initiation sites of human genes as a cause of inherited disease. Hum. Mutat. 32, 1137–1143 (2011).
Kline-Fath B., R.B.-S., Bulas D. Fundamental and Advanced Fetal Imaging: Ultrasound and MRI, (Wolters Kluwer, Chicago, IL, USA, 2015).
Acknowledgements
We would like to thank the family for their participation in our study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, Z., Schonberg, R., Guidugli, L. et al. A novel mutation in the promoter of RARS2 causes pontocerebellar hypoplasia in two siblings. J Hum Genet 60, 363–369 (2015). https://doi.org/10.1038/jhg.2015.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.31
This article is cited by
-
A non-coding variant in the Kozak sequence of RARS2 strongly decreases protein levels and causes pontocerebellar hypoplasia
BMC Medical Genomics (2023)
-
What’s new in pontocerebellar hypoplasia? An update on genes and subtypes
Orphanet Journal of Rare Diseases (2018)
-
Novel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia – further expansion of the phenotypic spectrum
Orphanet Journal of Rare Diseases (2016)
-
Structural basis for early-onset neurological disorders caused by mutations in human selenocysteine synthase
Scientific Reports (2016)
-
Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability
Metabolic Brain Disease (2016)
