
Abstract
Although extraocular muscles are commonly affected by myasthenia gravis (MG) at presentation, a treatment-resistant ophthalmoplegic complication of MG (OP-MG) occurs in younger patients with African-genetic ancestry. In MG, pathogenic antibodies activate complement-mediated muscle damage and this may be potentiated in some OP-MG cases because of relative deficiency of decay-accelerating factor/CD55. Extending this argument, we hypothesized that OP-MG individuals may harbor African-specific polymorphisms in key genes influencing extraocular muscle remodeling. We screened the regulatory region of the transforming growth factor beta-1 (TGFB1) gene encoding the cytokine pivotal in muscle healing responses. We show the frequency of an African-specific polymorphism TGFB1 c.−387 T (rs11466316) among South Africans with African-genetic ancestry is higher than 1000 Genomes African controls (17.2% vs 4.8%; P<1 × 10−7), and associates with juvenile OP-MG (28%; P=0.043). Further, TGFB1 −387 C>T is functional because it represses the TGFB1 promoter construct basal activity by fivefold, and OP-MG fibroblasts (−387 C/T or T/T) have lower basal TGFB1 mRNA transcripts compared with controls (−387 C/C)(P=0.001). Co-transfections with Sp1 show less responsiveness of the −387 T promoter compared with wild-type −387 C (P=0.015). Our findings suggest that population-specific alleles may lower TGFB1 expression, thereby influencing OP-MG susceptibility by inhibiting extraocular muscle CD55 upregulation and/or altered endplate remodeling.
Similar content being viewed by others

Introduction
Myasthenia gravis (MG) is an antibody-mediated disease characterized by fatigable weakness of the skeletal muscle. Pathogenic antibodies most frequently target the muscle endplate acetylcholine receptors (AChRs) resulting in complement-induced damage. Extraocular muscles are commonly affected as an early manifestation of generalized MG, but respond to treatment.1 We have identified individuals with African-genetic ancestry, commonly juveniles, who develop treatment-resistant ophthalmoplegia and ptosis (OP-MG).2, 3 Indeed, ~20% of juvenile AChR antibody positive MG subjects with African-genetic ancestry develop this complication regardless of whether the MG was confined to the ocular muscles or generalized.3
Complement-mediated endplate damage is a critical effector mechanism in AChR antibody positive MG.4, 5, 6, 7 When genes of specific complement regulatory proteins such as decay-accelerating factor (DAF) and/or CD59 were knocked down in an experimental allergic MG animal models, mice with no/lower DAF and/or CD59 expression suffered more severe complement-mediated endplate damage compared with their wild-type littermates.6, 8, 9, 10, 11, 12 In extraocular muscles compared with other muscles, the role of complement regulatory proteins may be more critical and therefore they are more susceptible to complement-mediated damage because of (a) lower basal and experimental allergic MG-induced expression of DAF1, 9, 13 and (b) possibly increased MG-mediated inflammation in the extraocular muscles.14
Previously, we reported that a functional polymorphism (−198 C>G) in the regulatory region of DAF associated with a subset of African OP-MG subjects.15 Although a larger proportion of MG subjects presented with ophthalmoplegia and ptosis as part of their initial MG symptom complex, most of the individuals with this DAF promoter variant developed the ophthalmoplegia only after MG was diagnosed and treatment initiated.16 We postulated that in the extraocular muscles, at critical periods, inadequate DAF upregulation and consequently complement protection increased the susceptibility to developing MG-associated ophthalmoplegia. As the OP-MG phenotype is likely multifactorial and not completely explained by DAF −198 C>G, we extended the ‘complement hypothesis’ to argue that in the context of autoimmune MG, muscle endplate damage will result in altered muscle repair pathways.
The cytokine, transforming growth factor beta-1 (TGF-β1), has a pivotal role in the response of muscle cells to injury; damaged muscle endplate as well as inflammatory cells in/around the muscle upregulate TGF-β1 expression.17 An autoinduction pathway in the muscle allows TGF-β1 expression to influence satellite cells to transdifferentiate into myofibroblasts or myoblasts.17, 18 In addition, TGF-β1 may have a marked effect on the contractile properties of the muscle,19 and the TGF-β1 signaling pathway shows differential regulation with higher transcript levels (and downstream SMAD4 protein levels) in a mouse extraocular muscle cell line compared with limb muscle cells.20
In a study examining the genetic diversity in the TGFB1 promoter, the serendipitous inclusion of an isolated South African individual with African-genetic ancestry revealed various novel single-nucleotide polymorphisms (SNPs).21 Furthermore, some of these African TGFB1 promoter polymorphisms were predicted to alter TGF-β1 expression.21 Owing to the role of TGF-β1 in injured muscle, we proposed that complement-mediated MG damage in individuals harboring African-specific TGFB1 promoter functional polymorphisms, may alter remodeling of extraocular muscles by impacting myogenesis, exhaustion of the resident satellite cell population and influencing myotube force-generating capacity. Here we show an association with OP-MG and an African-specific SNP found within the extended TGFB1 regulatory region, and demonstrate the functional implications of the African haplotype harboring this SNP.
Materials and methods
Patients
We previously reported the clinical characteristics of the OP-MG phenotype.2, 3 Briefly, we included two groups of OP-MG patients, which were defined as (1) complete OP-MG in which all 12 EOMs (frequently including eyelids with bilateral ptosis) show either no movement or only brief microsaccades (75−100% weakness), and (2) those with severe partial OP-MG where >50% of the EOMs show loss of motility (75–100% weakness). As MG controls, we included well-characterized patients followed for many years in the clinic who had not developed the OP-MG phenotype. All OP-MG patients had the classical features of MG, including fatigable weakness and pyridostigmine responsiveness, as well as improvement on immune therapies in non-ocular muscles. Of the OP-MG individuals, 23 of 26 (89%) had detectable AChR antibodies, 2 had MuSK antibodies and in 1 the radioimmunoassay was negative to both AChR and MuSK antibodies (Table 1). Individuals developing symptoms of MG before the age of 20 years were classified as juvenile MG.3, 22 Population controls were derived from normal controls who previously consented to participation in MG-related studies and categorized themselves according to South African census racial categories.15 The Sowetan data set raw values related to TGFB1, representing independent South African population controls, were kindly shared by the Bioinformatics unit, University of the Witwatersrand.23
The study was approved by the University of Cape Town Health Sciences Faculty Research Ethics committee and all individuals (or their parents if <18 years) signed informed consent to participate.
Cloning and sequencing of TGFB1 regulatory region
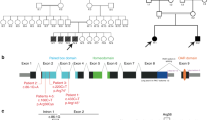
DNA was extracted from peripheral blood samples as described previously.15 The upstream regulatory region of TGFB1 including exon 1 and the 5′-untranslated region (5′-UTR) was sequenced in MG patients (n=78) and South African controls with African-genetic ancestry (n=65)15 using a set of sequencing primers spanning 10 polymorphic sites (Table 1). These 10 polymorphisms, shown in Figure 1, were selected for genotyping based on previous data implicating them in TGFB1 gene regulation or their unique occurrence in subjects of African-genetic ancestry (rs11466314, rs35318502, rs11466316, rs9282871).21 A 3193 bp fragment of the TGFB1 promoter was amplified using 5′-ACATGGCCTACTCCCTTCCT-3′ (forward) and 5′-CCTCCTTGGCGTAGTAGTCG-3′ (reverse) primers in a PCR reaction using 50 ng genomic DNA as template and Takara EmeraldAmp Max PCR Master Mix (Clontech Laboratories, Mountain View, CA, USA). As this reaction could not be sufficiently optimized to yield a product suitable for direct sequencing, 1 μl was then used as a template in a subsequent PCR reaction using internal primers to generate a 2880 bp product. These nested primers (forward, 5′-CCAGCTGAGCTCTCCCTCACAGCAATTA-3′ and reverse, 5′-GCTCCAAAGCTTTAGTCTTGCAGGTGGATAG-3′) included a SacI and HindIII restriction enzyme site, respectively (mismatches to the reference sequence in bold) for downstream cloning of the PCR TGFB1 promoter product. For both reactions, the following cycling conditions were used: 98 °C for 10 s (initial denaturation), 30 cycles of 98 °C for 10 s (denaturation), 62.2 °C for 30 s (annealing) and 72 °C for 3 min 30 s (extension), followed by 72 °C for 10 min (final extension) in a MultiGene Thermal Cycler (Labnet International, Edison, NJ, USA). Sanger sequencing reactions were performed on the final purified PCR product using various sequencing primers (shown in Table 1) using the ABI V3.1 Big Dye Kit on the ABI 3500XL machine (both Life Technologies, Carlsbad, CA, USA) according to the manufacturer specifications. Sequences were aligned to the reference sequence using CLC Main Workbench version 7.5.1 software (CLC Bio, Qiagen, Venlo, The Netherlands) to determine polymorphic alleles.

Schematic of the human-extended TGFB1 (transforming growth factor beta-1) promoter defining the regulatory regions and the 10 polymorphisms genotyped in this study. Diagram (a) compares the extended TGFB1 promoter reporter construct used in this study with other common TGFB1 promoter reporter constructs in the literature (b and c).
Haplotype analysis
Haploview software24 was used to perform haplotype analyses on the unphased genotyping data by applying the confidence interval (nearest-neighbor) algorithm described by Gabriel et al.25 As an additional analysis, the default confidence bound cutoffs were customized to only include −2389, −1347 and −387 polymorphic sites. Individuals with >50% missing marker genotypes were excluded from the analysis and pairwise comparisons of markers >500 kb apart were ignored.
TGFB1 promoter construct
To examine the functional effects of the TGFB1 regulatory region, we constructed a TGFB1 promoter reporter construct spanning the extended regulatory region by cloning a 2856 bp TGFB1 promoter fragment upstream of a luciferase reporter gene (Figure 1a). As the cloned TGFB1 promoter fragment was amplified from genomic DNA, it was sequenced to determine which polymorphic variants were present. The cloned fragment corresponded with nucleotides–2750 to +106 numbered relative to the ATG translation start site and is identical in sequence identity to the NCBI reference sequence NC_000019.10 from nucleotide positions 41 352 723 to 41355 915 (chromosome 19, GRCh38.p2 primary assembly 12-03-15), containing c.−1347 T and designated pHap-4.
Plasmid constructs
The pRL-TK vector (Promega, Madison, WI, USA) was used as an internal control reporter to test for transfection efficiency. The pCMV-Sp1 expression vector was obtained from Dr Tijan (Department of Molecular and Cell Biology, University of California, Berkeley, CA, USA) through Addgene (Cambridge, MA, USA). To create the various TGFB1 promoter haplotype constructs, −2389 delAGG, −1347 C and −387 T were introduced into the cloned TGFB1 promoter luciferase reporter construct template (pHap-4) by site-directed mutagenesis using the QuickChange Kit (Stratagene, La Jolla, CA, USA). This was carried out sequentially to generate pHap-1 to pHap-3 as shown in Figure 3b. Each promoter haplotype is biologically relevant (based on linkage disequilibrium data from our genotyping study) and was sequence verified to ensure that the desired mutation had been incorporated.
Sera collection
Sera from newly diagnosed AChR antibody positive MG patients with extraocular muscle involvement were collected in BD Vacutainer SST II Plus plastic serum tubes (Becton, Dickinson and Company, Franklin Lakes, NJ, USA). These samples were left to clot at room temperature for 4 h and centrifuged in a swinging bucket rotor centrifuge at 1900 g for 10 min at 4 °C. Serum was then removed, aliquoted and stored at −20 °C. Importantly, the patients selected for sera donation had generalized MG with substantial extraocular muscle involvement and were treatment-naïve (newly diagnosed or one individual known with MG but defaulted treatment for ⩾5 months). Control sera were collected from normal controls.
Primary fibroblast culture
Primary dermal fibroblasts were established from skin punch biopsies taken from the scapular region of OP-MG patients and MG controls using the explant method. Briefly, these biopsies were minced with sterile blades and cultured under coverslips in 6-well plates until the emergence of fibroblasts at 4–7 days. Once confluent, they were subcultured (2–3 weeks) and cryopreserved with 10% dimethyl sulfoxide at passage 2 in liquid nitrogen. Before cryopreservation, each fibroblast cell line was stained with Hoechst nuclear stain and screened for mycoplasma contamination using a fluorescent microscope.
Cell culture
HT1080 human fibrosarcoma (ATCC; CCL-121, Manassas, VA, USA) and the C2C12 mouse myoblast cell lines were maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal calf serum (Gibco, St Louis, MO, USA) and 1% penicillin–streptomycin at 37 °C (95% air, 5% CO2, 65% humidity). C2C12 cells were allowed to differentiate by switching growth medium to differentiating medium (Dulbecco’s modified Eagle’s medium supplemented with 2% horse serum and 1% penicillin–streptomycin). C2C12 cells were harvested at different differentiating time points.
Transfections and luciferase reporter assays
A day before transfection, cells were plated at 7.5 × 104 (HT1080) or 5.0 × 104 (C2C12) cells per ml in 12-well plates. Transient transfections were carried out using XtremeGene (Roche, Basel, Switzerland) according to the manufacturer’s instructions with DNA consisting of TGFB1 promoter haplotype constructs (150 ng for HT1080 and 1 μg for C2C12) together with 50 ng pRL-TK internal control vector. At 30 h after transfection, cells were harvested and analyzed for luciferase activity using the Dual-Luciferase Reporter Assay (Promega) following the manufacturer’s instructions and quantified using a Luminoskan Ascent luminometer (Thermo LabSystems, Franklin, MA, USA). All transfections were carried out in duplicate. For Sp1 experiments, HT1080 cells were co-transfected with 10–50 ng of a CMV-Sp1 expression vector. For stimulation experiments, C2C12 cells were transfected for 6 h and then serum-starved and treated with 5 mm sodium butyrate (NaB) (Sigma-Aldrich, St Louis, MO, USA) for 6 h.
Quantitative real-time PCR
RNA was extracted from confluent 6 cm dishes of unstimulated OP-MG and control dermal fibroblasts from donors <30 years of age using the HighPure RNA Extraction Kit (Roche). One microgram of RNA was reverse transcribed to cDNA using the ImPromII Reverse Transcription Kit (Promega). Quantitative PCR was performed on the cDNA samples using the Kapa SYBR FAST qPCR Master Mix (Kapa Biosystems, Wilmington, MA, USA) and primers for TGFB1 (Quantitect primer assay; Qiagen) in glass capillaries on the Roche LightCycler 2.0 instrument (Roche). A standard curve for TGFB1 was constructed by plotting the CT values generated by serial dilutions of a purified TGFB1 PCR product as template and this graph equation was used to quantify the fibroblast TGFB1 mRNA levels in terms of absolute copy number.
Western blot analysis
Following serum starvation for 24 h, subconfluent fibroblast cultures were either left untreated or stimulated with recombinant human TGF-β1 (rhTGF-β1) at a concentration of 5 ng ml−1 (R&D Systems, Minneapolis, MN, USA) for 60 min. Following this, cells were rinsed with ice-cold phosphate-buffered saline solution and then homogenized in RIPA lysis buffer containing protease and phosphatase inhibitors. Equal amounts of protein lysates (35 or 40 μg) were separated on 10% sodium dodecyl sulfate–polyacrylmide gels and transferred to nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). The membranes were probed with appropriate primary antibodies detected using peroxidase-conjugated anti-mouse or anti-rabbit antibodies and visualized by advanced chemiluminescence (Pierce, Rockford, IL, USA). The primary antibodies used were rabbit monoclonal anti-phospho-Smad3 (C25A9; Cell Signalling, Beverly, MA, USA) and rabbit polyclonal anti-p38 (Cell Signalling Technology, Berverly, MA, USA). The densities of the bands on the developed film were analyzed using UN-SCAN-IT gel software (version 6.1, Silk Scientific, Inc., Orem, UT, USA).
Statistical analysis
The clinical data are skewed and represented by median and interquartile ranges. For the genotyping data, allele and genotype frequencies were compared between groups using 2 × 2 tables (http://www.openepi.com) and proportional analyses were performed using Mid-P Exact test with the conditional maximum likelihood estimate of odds ratio (OR) and 95% confidence interval (CI). ORs were not calculated for values equal to zero. The Bonferroni correction was not applied owing to the background linkage disequilibrium that exists in the regulatory region of TGFB1.26 Comparative statistics on haplotype frequencies between groups was performed in Haploview using χ2 tests. Continuous, normally distributed data sets generated from luciferase assays and quantitative PCR (small sample size) were appropriately compared using Student’s unpaired two-sample t-test (Excel). For all analyses, P⩽0.05 (two-tailed) was considered significant. Graphs were generated in the GraphPad software (Prism 6 for Mac OS × version 6.0).
Results
Subjects
We genotyped a total of 78 MG patients with African-genetic ancestry: 26 OP-MG individuals (complete and severe partial ophthalmoplegia) and 52 control MG individuals who have had MG for ⩾3 years without developing persistant extraocular muscle involvement (median follow-up 14 years, interquartile range 13.5–19 years). Table 2 shows the clinical characteristics of the cohort. Individuals categorised themselves according to South African census racial categories: 30 were indigenous African/black individuals and 48 were mixed ancestry (as described previously).2 The MG sample included 23 men and 55 women. The normal South African controls included 33 indigenous African/black individuals and 32 of mixed African-genetic ancestry (mixed ancestry).
Genotyping of TGFB1 regulatory region
We sequenced the regulatory region of the TGFB1 gene spanning −2750 to +106 to genotype 10 polymorphisms, including rare SNPs that have only been reported in subjects with African-genetic ancestry (Figure 1a). For comparison, Figures 1b and c show the TGFB1 promoters previously used for association and functional studies which do not include two African-specific SNPs in the 5′-UTR. Table 3 shows the associations of allele and genotype frequencies of the 10 polymorphisms in 52 chromosomes of all OP-MG individuals (complete and partial severe), compared with control MG (104 chromosomes) who have had MG for ⩾3 years. The minor allele −1347 T (rs1800469) shows a trend towards a ‘protective’ association with OP-MG (OR=0.4; P=0.071) and was not found in the juvenile OP-MG sample despite being present in 20% of MG controls (P=0.038). In contrast, two rare 5′-UTR African SNPs (haplotype block 2) associate with juvenile onset complete OP-MG: −387C>T (rs11466316; OR=3.9; P=0.043; 95% CI: 1.0, 13.7) and −14G>A (rs9282871 OR=4.2; P=0.038; 95% CI 1.1, 15.0).
The OP-MG group with −387 C/T genotype (n=7) had a mean age at symptom onset of 14.6 years vs the −387 C/C genotype (n=19), with a mean age of 22.6 years (P=0.20). The MG controls with −387 C/T (n=9) vs C/C genotypes (n=42) had mean ages at MG onset of 29.4 vs 27.3 years, respectively (P=0.08).
The 10 TGFB1 polymorphisms genotyped in our MG sample are in strong linkage disequilibrium and inherited as sets of closely linked markers in two haplotype blocks (Figure 2a): block 1 extends across the enhancer and negative regulatory regions and comprises four common SNPs including the ‘protective’ rs1800469; block 2 contains rarer African-specific polymorphisms in the 5′-UTR including the two African-specific variants that associate with juvenile complete OP-MG (rs11466316 and rs9282871).

Haplotype block analysis and minor allele frequencies of TGFB1 (transforming growth factor beta-1) promoter polymorphisms. (a) Certain TGFB1 promoter polymorphisms genotyped in our myasthenia gravis (MG) sample are in strong linkage disequilibrium (LD) and are inherited as sets of closely linked markers in two haplotype blocks: block 1 containing common polymorphisms spanning the two enhancer and negative regulatory regions and block 2 containing rarer African-specific polymorphisms in the 5′-untranslated region (5'-UTR) (LD plot showing R2 values and haplotype block analysis performed in Haploview). (b) Minor allele frequency data for the 10 selected TGFB1 promoter single-nucleotide polymorphisms (SNPs) in EUR (1000 Genomes European sample), AFR (1000 Genomes African sample) and SA AFR MG (South African MG sample). The dotted line at 5% indicates the frequency below which alleles are considered to be rare variants. *P<0.05, **P<0.005.
The African-specific TGFB1 −387C>T promoter variant (rs11466316) is a common polymorphism in South Africans
By comparing the minor allele frequency data for our total MG sample (n=156 chromosomes) with the European (n=1006) and ‘African’ (n=1322) samples in the 1000 Genomes data set (Figure 2b), we show that, while our South African MG cohort is predictably different to the European sample, they also differ significantly from the ‘Africans’ comprising pooled African-American and East- and West-African populations. Notably, rs11466316 (−387 C>T) is absent in Europeans, present as a rare variant in the 1000 Genomes African data set (minor allele frequency <0.05), and not uncommon as a variant in our total South African MG sample with African-genetic ancestry present at a minor allele frequency of 11% (AFR 1000G vs SA MG, P=0.006). Furthermore, this does not appear to be peculiar to our MG group as the TGFB1 −387 T allele is present at a frequency of 16% and 18% in two independent South African population control cohorts from Cape Town (black and mixed ancestry; n=130, our data) and a previously reported black cohort from Soweto (n=188), respectively.23 Combined (n=318 chromosomes) they differ significantly from the 1000 Genomes African data set (n=1322; P=1 × 10−7; Figure 3a).

TGFB1 (transforming growth factor beta-1) −387 T (rs11466316) allele frequencies among South Africans with African-genetic ancestry. (a) The −387 C>T (rs11466316) polymorphism is a common African-specific variant in South Africans. Minor allele frequency data for −387 T in African controls is presented from various sources: 1322 African Americans and East and West Africans (1000 Genomes Database), 65 individuals from Cape Town, South Africa (genotyped in this study) and 94 individuals from Soweto, South Africa.23 (b) pHap-3, containing the African-specific −387 T allele, is more common in juvenile treatment-resistant ophthalmoplegic complication (OP-MG) patients (28%) compared with MG controls (9%), P=0.02.
TGFB1 pHap-3 containing the −387 T allele associates with juvenile onset OP-MG
We performed a haplotype analysis of the following three polymorphic loci: (a) c.−387 T because it associates with juvenile onset OP-MG and in silico analysis predicts that it may influence TGFB1 transcriptional activity and (b) two closely linked SNPs in the distal regulatory region (rs11466313 and rs1800469) because they are under-represented in OP-MG compared with MG controls and reported to be functional.27, 28 Subjects in our MG group had one of four possible TGFB1 promoter haplotypes (pHap-1 to pHap-4) as shown in Figure 3b. Indeed, the identified protective and susceptibility alleles segregate; in pHap-2 the ‘protective’ −1347 T allele is more frequent in MG controls and pHap-3 containing the −387 T allele is more frequent in juvenile OP-MG cases (28%) compared with MG controls (9%; P=0.023; Figure 3b and Table 3).
The African TGFB1 −387 C>T variant is functional
We next determined if the −387 T allele impacts on TGFB1 promoter activity because it is associated with juvenile OP-MG and has been reported to alter Sp1 and Sp3 DNA binding.21 To this end, luciferase promoter reporter constructs of pHap-1 to pHap-4 were transiently transfected into HT1080 human fibrosarcoma cells and C2C12 mouse myoblast cells and luciferase activity measured. The most striking finding was that pHap-3, which contains the African-specific −387 T allele, showed a loss of basal promoter activity relative to the other three haplotypes (Figure 4). Importantly, pHap-3 consistently showed >5-fold reduction in basal TGFB1 promoter activity when compared with p-Hap-1 in both cell lines tested (P<0.0002). As p-Hap-1 and pHap-3 differ only at position −387, one can directly attribute this difference in promoter activity to the −387 T polymorphism.

pHap-3 containing the African-specific −387 T allele reduces basal TGFB1 (transforming growth factor beta-1) transcriptional activity. HT1080 cells (a) and C2C12 cells (b) were transiently transfected with pGL3 luciferase reporter gene constructs containing TGFB1 promoter haplotypes pHap-1 to pHap-4 (generated using site-directed mutagenesis) together with TK renilla reporter gene construct as an internal control for transfection efficiency. Thirty hours following transfection, cells were lysed and luciferase activity measured. The ratio of firefly to Renilla luciferase activity was normalized against the empty promoterless pGL3-basic vector in each cell line. Fold activation for each promoter haplotype relative to the empty vector is shown and statistically significant differences in fold activation between the various promoter haplotypes are indicated. These are pooled results of ⩾3 independent experiments, each performed in duplicate. Student's unpaired t-test, two-tailed; errors bars s.e.m.
It is worth noting that while the −14 A allele (rs9282871) is in strong linkage disequilibrium with −387 T (R2=0.86), it had no reported effect on nuclear factor binding21 and our promoter studies examining this variant (pHap-1 mutated at −14 G>A) showed no difference in reporter gene expression (Supplementary Figure 1). Thus, we concluded that this SNP does not influence TGFB1 promoter activity and excluded it from further analysis.
The TGFB1 promoter contains several putative Sp1 binding sites (GC boxes) and is indeed transcriptionally activated by Sp1. The sequence surrounding the −387 C>T polymorphism is a partial Sp1 binding site and the −387 T allele alters this motif and was reported to abrogate Sp1 binding.21 However, whether this affects TGFB1 promoter activity was not investigated. We therefore determined the functional significance of −387 T allele on TGFB1 promoter responsiveness to Sp1 in luciferase reporter assays. HT1080 cells were co-transfected with pHap-1 and pHap-3 luciferase reporters together with increasing concentrations of a Sp1 expression vector. We show that compared with pHap-1, pHap-3 is less responsive to stimulation by Sp1 (P=0.015; Figure 5). This indicates that the partial Sp1 site surrounding the −387 C>T polymorphism is functional and that it has an important role in the regulation of the TGFB1 promoter by Sp1.

TGFB1 (transforming growth factor beta-1) −387 C>T (rs11466316) shows differential sensitivity to Sp1. To investigate whether the −387 C>T polymorphism alters the sensitivity of the TGFB1 promoter to Sp1 activation, HT1080 cells were transiently transfected with pGL3 luciferase reporter gene constructs containing TGFB1 promoter haplotypes pHap-1 and pHap-3 together with a cytomegalovirus (CMV) empty vector and incremental doses of a CMV-Sp1 expression vector. Thirty hours after transfection, cells were lysed and luciferase activity measured. The ratio of firefly to Renilla luciferase activity for each Sp1 dose was normalized against the CMV empty vector for each promoter haplotype. Fold activation for each promoter haplotype relative to the CMV empty vector are shown and the 50ng Sp1 dose, which showed significant differences in fold activation between pHap-1 and pHap-3 is indicated. These are pooled results of ⩾3 independent experiments, each performed in duplicate. Student's unpaired t-test, two-tailed; errors bars s.e.m.
NaB is a histone deacetylase inhibitor that stimulates the promoters of several genes through Sp1/Sp3 binding sites.29, 30 In vitro studies have also shown the TGFB1 promoter to be activated in response to NaB treatment, but whether this involves Sp1 has not been demonstrated.31, 32 We therefore assessed whether the −387 T allele alters the response of the TGFB1 promoter to NaB. To this end, C2C12 cells transfected with pHap-1 and pHap-3 promoter constructs were treated with 5 mm NaB for 6 h and luciferase reporter activity measured. In contrast to other reports, our results show that NaB greatly represses both pHap-1 and pHap-3 promoter constructs to the same degree (100-fold; P⩽1 × 10−6) (Supplementary Figure 2). This suggests that the −387 SNP is not important in regulating the response of the TGFB1 promoter to NaB and that the promoter used in our study may contain additional regulatory elements not contained in the TGFB1 promoters used in other studies involving NaB.
TGFB1 −387 C>T is an expression quantitative trait in phenotype-specific dermal fibroblasts
We next investigated whether the observed differences in promoter activity of pHap-1 and pHap-3 translate into altered TGFB1 expression levels in phenotype-specific dermal fibroblasts. To this end, we extracted RNA from low passage dermal fibroblast cell lines derived from OP-MG (−387 C/T or T/T genotype) and control MG patients (−387 C/C genotype) and measured basal TGFB1 transcripts by quantitative PCR. Consistent with our promoter studies, we found that fibroblasts with the −387 C/C genotype have >2-fold higher TGFB1 mRNA transcript abundance than those with the −387 C/T or T/T genotype (P=0.001) (Figure 6a). This suggests that, at the very least in fibroblasts, the TGFB1 −387 promoter genotype strongly predicts TGFB1 mRNA levels.

TGFB1 (transforming growth factor beta-1) −387 C>T is an expression quantitative trait in phenotype-specific dermal fibroblasts. (a) Treatment-resistant ophthalmoplegic complication (OP-MG) dermal fibroblasts (−387 C/T or T/T genotype) have lower TGFB1 expression levels compared with MG control dermal fibroblasts (−387 C/C). Low passage fibroblast cell lines were cultured from skin biopsies from OP-MG (−387 C/T or T/T, n=3 individuals) and MG control patients (−387 C/C, n=2 individuals). RNA was harvested and equal quantities from each cell line were reverse transcribed to cDNA. Quantitative PCR (qPCR) was performed on each cDNA sample using primers for TGFB1. TGFB1 mRNA expression levels were determined using absolute quantification by interpolating CT values from a TGFB1 standard curve (generated using serial dilutions of a purified TGFB1 PCR product as an external standard). Each sample was tested in duplicate and in the same qPCR run and results are pooled to compare TGFB1 expression levels between the two genotype groups for the −387 C>T polymorphism. Student’s unpaired t-test, two-tailed; error bars s.e.m. (b) Short duration (60 min) recombinant human TGF-β1 (rhTGF-β1) (5 ng ml−1) stimulation shows repression of phosporylated Smad3 levels in skin-derived fibroblasts from subjects with OP-MG (−387 C/T genotype) compared with those with control MG (−387 C/C genotype). Pooled densitometric readings from western blots of control MG (n=3 blots, three individuals) and OP-MG fibroblasts (n=5 blots from three individuals) in response to 5ng rhTGF-β1 stimulation (60 min) are shown. Phosphorylated Smad3 levels are used as a measure of the downstream effect of rhTGF-β1 stimulation normalized to p38 expression. Results are expressed as fold change relative to untreated cells.
While genetic variants influencing gene expression are frequently cell type-specific, mimicking physiological conditions may inform their directional function; that is, activation or repression on cell type-specific pathways.33 Based on the above results, we speculated that in response to stimulating the autocrine TGF-β1 pathway with rhTGF-β1,18 control fibroblasts with the −387 C/C genotype will have higher TGF-β1 expression levels compared with OP-MG fibroblasts with the −387 C/T genotype. To test this, we determined levels of phosphorylated Smad3 (p-Smad3), a marker of the canonical TGF-β1 signaling pathway, by western blotting. In keeping with the promoter and quantitative PCR results, densitometric levels of p-Smad3 in −387 C/T fibroblasts were significantly lower compared with −387 C/C fibroblasts (P=0.015) (Figure 6b and Supplementary Figure 3).
Discussion
We previously reported that individuals with AChR antibody positive MG and African-genetic ancestry, and particularly those developing MG before the age of 20 years (juvenile MG), have a significantly higher risk of developing treatment-resistant ophthalmoplegia.2, 3, 22 Here we show that juvenile myasthenics who develop severe/complete ophthalmoplegia have fourfold higher odds of harboring an African-specific functional TGFB1 c.−387 T containing haplotype, compared with racially appropriate MG controls without the ophthalmoplegic complication. The TGFB1 −387 C>T polymorphism was previously reported to alter Sp1 and Sp3 DNA binding.21 We find that TGFB1 −387 T results in lower basal TGFB1 promoter activity in a fibroblast and muscle cell line, and that this repressive response is likely related to altered Sp1 sensitivity. In addition, fibroblasts from MG patients with TGFB1 −387 T alleles (T/T or C/T genotypes) have significantly lower basal TGFB1 transcripts compared with −387 C/C haplotypes. These findings suggest that TGFB1 −387 C>T is an expression quantitative trait locus for the development of the ophthalmoplegic complication observed in MG patients with African-genetic ancestry.
Here we describe several important observations concerning our population genotyping data including that (1) the African-specific haplotype (p-Hap3) was over-represented in OP-MG patients and under-represented in MG patients who do not develop persistent ophthalmoplegia and (2) databases such as the 1000 Genomes Project are not representative of African genomes from Southern Africa, which are increasingly recognized as being more diverse owing to their ancient origins.34, 35 This may also explain why the ophthalmoplegic MG complication is much less frequently recognized among those with African-genetic ancestry elsewhere in the world (unpublished communications).
We also present novel data using an extended TGFB1 promoter reporter construct that includes many more regulatory elements relative to constructs used in other reported studies, such as the second promoter and 5′-UTR and the most distal enhancer region.21, 27(Figure 1). This could clarify why we observed NaB induced repression of the TGFB1 promoter, which conflicted with previously published promoter studies. The TGFB1 promoter has a partial NaB response element in a negative regulatory region (positions −1333 to −1323), similar to that found in the cyclin D1 promoter, which is repressed by NaB.36 Indeed, our TGFB1 promoter construct may more accurately reflect the endogenous regulatory region of the TGFB1 gene as our results are consistent with data showing that butyrate treatment decreased endogenous TGF-β1 levels in renal epithelial cells.37 Furthermore, our TGFB1 promoter construct allows the examination of the combined effect of various SNPs throughout the regulatory region in a biologically relevant manner.
In conclusion, our results reveal an African-specific SNP in the TGFB1 gene, which reduces TGFB1 levels leading to a reduction of its downstream canonical signaling pathway. This SNP associates with juvenile OP-MG and our results therefore have implications for our understanding of the pathogenesis of this complication (see Figure 7). Evidence is accumulating that the susceptibility of extraocular muscle to MG-associated complement damage is substantially higher compared with other skeletal muscle; extraocular muscles have relatively lower DAF expression levels,13 and in addition to MG-induced DAF downregulation, these muscles may be susceptible to greater inflammatory cell infiltrates,14 all of which are expected to increase complement damage. In human orbital fibroblasts, TGF-β1 has been shown to upregulate DAF expression, which likely protects against complement damage during inflammation.38 The uniquely African TGFB1 promoter haplotype described in this report may also confer 'high risk' for developing OP-MG as it is associated with lower TGFB1 transcripts and possibly less DAF induction in the orbital microenvironment. These factors may also impact on adaptive immune responses in the orbital microenvironment.12 Therefore, we postulate that critically low extraocular muscle DAF expression levels, either due to MG-induced DAF repression (susceptible subjects with DAF −198 C>G)15 and/or low TGF-β1/Smad3 canonical signaling, contribute to the manifestation of the ophthalmoplegic complication in juvenile MG.

A model of the relationships between African-specific genetic susceptibility variants and the development of the ophthalmoplegic complication in myasthenia gravis. AChR, acetylcholine receptor; DAF, decay-accelerating factor; EOM, extraocular muscle; MG, myasthenia gravis; TGFB1, transforming growth factor beta-1.
References
Soltys, J., Gong, B., Kaminski, H. J., Zhou, Y. & Kusner, L. L. Extraocular muscle susceptibility to myasthenia gravis: unique immunological environment? Ann. NY Acad. Sci. 1132, 220–224 (2008).
Heckmann, J. M., Owen, E. P. & Little, F. Myasthenia gravis in South Africans: racial differences in clinical manifestations. Neuromuscul. Disord. 17, 929–934 (2007).
Heckmann, J. M., Hansen, P., Van Toorn, R., Lubbe, E., Janse van Rensburg, E. & Wilmshurst, J. M. The characteristics of juvenile myasthenia gravis among South Africans. S. Afr. Med. J. 102, 532–536 (2012).
Biesecker, G. & Gomez, C. M. Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J. Immunol. 142, 2654–2659 (1989).
Lennon, V. A., Seybold, M. E., Lindstrom, J. M., Cochrane, C. & Ulevitch, R. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J. Exp. Med. 147, 973–983 (1978).
Morgan, B. P., Chamberlain-Banoub, J., Neal, J. W., Song, W., Mizuno, M. & Harris, C. L. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin. Exp. Immunol. 146, 294–302 (2006).
Sahashi, K., Engel, A. G., Lambert, E. H. & Howard, F. M. Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J. Neuropathol. Exp. Neurol. 39, 160–172 (1980).
Kaminski, H. J., Kusner, L. L., Richmonds, C., Medof, M. E. & Lin, F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp. Neurol. 202, 287–293 (2006).
Kaminski, H. J., Li, Z., Richmonds, C., Lin, F. & Medof, M. E. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp. Neurol. 189, 333–342 (2004).
Lin, F., Kaminski, H. J., Conti-Fine, B. M., Wang, W., Richmonds, C. & Medof, M. E. Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J. Clin. Invest. 110, 1269–1274 (2002).
Medof, M. E., Kinoshita, T. & Nussenzweig, V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J. Exp. Med. 160, 1558–1578 (1984).
Soltys, J., Halperin, J. & Xuebin, Q. DAF/CD55 and Protectin/CD59 modulate adaptive immunity and disease outcome in experimental autoimmune myasthenia gravis. J. Neuroimmunol. 244, 63–69 (2012).
Porter, J. D., Khanna, S., Kaminski, H. J., Rao, J. S., Merriam, P., Richmonds, C. R. et al. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc. Natl Acad. Sci. USA 98, 12062–12067 (2001).
Zhou, Y., Kaminski, H. J., Gong, B., Cheng, G., Feuerman, J. M. & Kusner, L. RNA expression analysis of passive transfer myasthenia supports extraocular muscle as a unique immunological environment. Invest. Ophthalmol. Vis. Sci. 55, 4348–4359 (2014).
Heckmann, J. M., Uwimpuhwe, H., Ballo, R., Kaur, M., Bajic, V. B. & Prince, S. A functional SNP in the regulatory region of the decay-accelerating factor gene associates with extraocular muscle pareses in myasthenia gravis. Genes Immun. 11, 1–10 (2010).
Auret, J., Abrahams, A., Prince, S. & Heckmann, J. M. The effects of prednisone and steroid-sparing agents on decay accelerating factor (CD55) expression: implications in myasthenia gravis. Neuromuscul. Disord. 24, 499–508 (2014).
Burks, T. N. & Cohn, R. D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 1, 1–13 (2011).
Li, Y., Foster, W., Deasy, B. M., Chan, Y., Prisk, V., Tang, Y. et al. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am. J. Pathol. 164, 1007–1019 (2004).
Mendias, C. L., Gumucio, J. P., Davis, M. E., Bromley, C. W., Davis, C. S. & Brooks, S. V. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 45, 55–59 (2012).
Porter, J. D., Israel, S., Gong, B., Merriam, A. P., Feuerman, J., Khanna, S. et al. Distinctive morphological and gene/protein expression signatures during myogenesis in novel cell lines from extraocular and hindlimb muscle. Physiol. Genomics 24, 264–275 (2006).
Shah, R., Rahaman, B., Hurley, C. K. & Posch, P. E. Allelic diversity in the TGFB1 regulatory region: characterization of novel functional single nucleotide polymorphisms. Hum. Genet. 119, 61–74 (2006).
Mombaur, B., Lesosky, M. R., Liebenberg, L., Vreede, H. & Heckmann, J. M. Incidence of acetylcholine receptor-antibody-positive myasthenia gravis in South Africa. Muscle Nerve 51, 533–537 (2015).
May, A., Hazelhurst, S., Li, Y., Norris, S., Govind, N., Tikly, M. et al. Genetic diversity in black South Africans from Soweto. BMC Genomics 14, 1–12 (2013).
Barrett, J. C, Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Nyholt, D. R. A simple correction for multiple testing for single-nucleotide polymorphisms in linage disequilibrium with each other. Am. J. Hum. Genet. 74, 765–769 (2004).
Healy, J., Dionne, J., Bélanger, H., Larivière, M., Beaulieu, P., Labuda, D. et al. Functional impact of sequence variation in the promoter region of TGFB1. Int. J. Cancer 125, 1483–1489 (2009).
Shah, R., Hurley, C. K. & Posch, P. E. A molecular mechanism for the differential regulation of TGF-beta1 expression due to the common SNP −509C-T (c. −1347C>T). Hum. Genet 120, 461–469 (2006).
Walker, G. E., Wilson, E. M., Powell, D. & Oh, Y. Butyrate, a histone deacetylase inhibitor, activates the human IGF binding protein-3 promoter in breast cancer cells: Molecular mechanism involves an Sp1/Sp3 multiprotein complex. Endocrinology 142, 3817–3827 (2001).
Yang, J., Kawai, Y., Hanson, R. W. & Arinze, I. J. Sodium butyrate induces transcription from the Gαi2 gene promoter through multiple Sp1 sites in the promoter and by activating the MEK-ERK signal transduction pathway. J. Biol. Chem. 276, 25742–25752 (2001).
Barnard, J. A. & Warwick, G. Butyrate rapidly induces growth inhibition and differentiation in HT-29 cells. Cell Growth Differ. 4, 495–501 (1993).
Skeen, V. R., Collard, T. J., Southern, S. L., Greenhough, A., Hague, A., Townsend, P. A et al. BAG-1 suppresses expression of the key regulatory cytokine transforming growth factor β (TGF-β1) in colorectal tumour cells. Oncogene 32, 4490–4499 (2012).
Knight, J. Approaches for establishing the function of regulatory genetic variants involved in disease. Genome Med 6, 1–15 (2014).
Schuster, S. C., Miller, W., Ratan, A., Tomsho, L. P., Giardine, B., Kasson, L. R. et al. Complete Khoisan and Bantu genomes from southern Africa. Nature 463, 943–947 (2010).
Tishkoff, S. A., Reed, F. A., Friedlaender, F. R., Ehret, C., Ranciaro, A., Froment, A. et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009).
Davie, J. R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 133, 2485 S–2493 S (2003).
Matsumoto, N., Riley, S., Fraser, D., Al-Assaf, S., Ishimura, E., Wolever, T. et al. Butyrate modulates TGF-β1 generation and function: potential renal benefit for Acacia(sen) SUPERGUMTM (gum arabic)? Kidney Int. 69, 257–265 (2006).
Cocuzzi, E. T., Bardenstein, D. S., Stavitsky, A., Sundarraj, N. & Medof, M. E. Upregulation of DAF (CD55) on orbital fibroblasts by cytokines. Differential effects of TNF-beta and TNF-alpha. Curr. Eye Res. 23, 86–92 (2001).
Acknowledgements
MN received a University of Cape Town Neurology fellowship and a South African Medical Research Grant (MRC) Post-Intern scholarship, JMH received support from the South African MRC, RR received a Discovery fellowship, J-MB a National Research Foundation (NRF) Scholarship, and SP received support from the MRC and NRF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Nel, M., Buys, JM., Rautenbach, R. et al. The African−387 C>T TGFB1 variant is functional and associates with the ophthalmoplegic complication in juvenile myasthenia gravis. J Hum Genet 61, 307–316 (2016). https://doi.org/10.1038/jhg.2015.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.146
This article is cited by
-
Gene expression profiling of orbital muscles in treatment-resistant ophthalmoplegic myasthenia gravis
Orphanet Journal of Rare Diseases (2020)
-
The functionality of African-specific variants in the TGFB1 regulatory region and their potential role in HIVAN
Clinical and Experimental Nephrology (2018)
-
RETRACTED ARTICLE: Juvenile-onset myasthenia gravis: autoantibody status, clinical characteristics and genetic polymorphisms
Journal of Neurology (2017)

{kind=link}
{kind=link}
{kind=link}