
Abstract
Recent progress in the study of hereditary large vessel diseases such as Marfan syndrome (MFS) have not only identified responsible genes but also provided better understanding of the pathophysiology and revealed possible new therapeutic targets. Genes identified for these diseases include FBN1, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, SKI, EFEMP2, COL3A1, FLNA, ACTA2, MYH11, MYLK and SLC2A10, as well as others. Their dysfunction disrupts the function of transforming growth factor-β (TGF-β) signaling pathways, as well as that of the extracellular matrix and smooth muscle contractile apparatus, resulting in progression of structural damage to large vessels, including aortic aneurysms and dissections. Notably, it has been shown that the TGF-β signaling pathway has a key role in the pathogenesis of MFS and related disorders, which may be important for development of strategies for medical and surgical treatment of thoracic aortic aneurysms and dissections.
Similar content being viewed by others


Introduction
Large vessel diseases, including aortic aneurysms and dissections, account for 1–2% of all deaths in Western countries.1 There are two types of aortic aneurysms based on their location: thoracic aortic aneurysm (TAA) and abdominal aortic aneurysm (AAA). The incidence of TAA and dissection (TAAD) is about 10 per 100 000 person-years.2 Approximately 20% of TAA patients have a positive family history while 10–20% of patients with AAA also have such a positive history. However, most patients with AAA also have various lifestyle-associated risk factors for cardiovascular events, such as smoking, hyperlipidemia, hypertension, sex and age.3 Therefore, AAA is thought to be a multifactorial disease that includes a genetic cause, with several genes and loci identified to have a positive association with the disease.
In this review, we focus on TAAD, which has a stronger genetic factor than AAA. Excessive matrix degradation is thought to be involved in the development of TAAD, though the exact pathological mechanisms are not yet fully understood. An imbalance between matrix metalloproteinases and their inhibitors is thought to induce medial degeneration and aneurysm development. In addition, some other conditions, including systemic arterial hypertension, can lead to thickening and fibrosis of the intimal layer and degradation and apoptosis of smooth muscle cells, resulting in wall weakness and eventually development of aneurysms, dissections and ruptures.
Over the past two decades after discovery of the FBN1 gene as a causative factor for Marfan syndrome (MFS), it has become clear that genetic factors are also at the basis of TAAD formation. Studies of the human syndromic form of TAAD, including MFS and its counterparts in mice, have provided a better general understanding of the condition, including the finding that the transforming growth factor-β (TGF-β) signaling pathway has a key role in the pathogenesis of TAAD. As the genes for several other syndromic aneurysmal conditions (Loeys-Dietz syndrome, arterial tortuosity syndrome, autosomal recessive cutis laxa, aortic osteoarthritis syndrome, others) have been identified, it is now considered that there is ample evidence showing that dysregulation of TGF-β signaling is the common final process in aortic diseases. In addition, recent findings have led to new therapeutic strategies for patients with TAAD.
MFS: syndromic TAAD and TGF-β signaling
MFS (OMIM #154700) is an autosomal-dominant connective tissue disorder first described by Antoine Marfan4 and with a prevalence of about 1 per 5000 individuals.5 The syndrome results from mutations in the FBN1 gene encoding fibrillin-1, a large glycoprotein and component of extracellular matrix microfibrils. MFS is a systemic disorder that affects skeletal (increased height, disproportionately long limbs and digits, anterior chest deformity, joint laxity, vertebral column deformity, narrow highly arched palate with crowding of teeth), ocular (myopia, increased axial globe length, corneal flatness, ectopia lentis) and cardiovascular (dilatation of aorta, especially aortic root, aortic regurgitation, mitral valve prolapse, mitral regurgitation) systems. Other phenotypes of MFS include dural ectasia, striae distensae, inguinal hernias, pneumothorax and pulmonary emphysema, though cardiovascular phenotypes, especially TAA, are the most important causes of morbidity and mortality in MFS patients. In the majority of cases, the primary dilatation occurs at the aortic root, especially at the level of the sinus of Valsalva.
For clinical diagnosis of MFS, first the Berlin,6 later the Ghent7 and most recently the revised Ghent nosology8 (Table 1) have been proposed. The revised Ghent nosology is thought to facilitate accurate recognition of MFS and differential diagnosis from other similar TAAD syndromes, thereby improving patient management as well as counseling. Four possible sets of findings can lead to a diagnosis of MFS in a proband: aortic root dilatation (Z score >2) and ectopia lentis, aortic root dilatation with an FBN1 mutation, aortic root dilatation with sufficient systemic findings (score of ⩾7 on systemic scale), and ectopia lentis with an FBN1 mutation that has been previously associated with aortic root dilatation. As compared with the previous version, the revised Ghent nosology adds more weight to genetic information, especially the relevant FBN1 mutation.
FBN1 and TGF-β signaling pathways
Fibrillin-1 regulates the signaling pathway of the TGF-β cytokine by interacting with latency-associated peptide (LAP), which is a protein derived from the N-terminal region of the TGF-β gene product and latent TGF-β-binding protein (LTBP)9 (Figure 1). After TGF-β is synthesized, it interacts with LAP to form a complex termed small latent complex, which is bound by LTBP to form a larger complex called large latent complex, and then secreted to the extracellular matrix. Thereafter, TGF-β remains in the extracellular matrix in an inactivated complex with LTBP and LAP, and this inactive complex regulates mediation of TGF-β signaling. As fibrillin-1 interacts with LTBP and LAP to regulate the level of TGF-β, its dysfunction results in activation of TGF-β signaling.10 It was initially thought that upregulation of TGF-β signaling occurred in canonical (SMAD-dependent) pathways. However, recent findings have shown changes in non-canonical (SMAD-independent) TGF-β pathways involving mitogen-activated protein kinases, including extracellular signal-regulated kinase 1/2, p38 and Jun N-terminal kinase. Therefore, increased TGF-β signaling via both canonical and non-canonical pathways contributes to aortic lesion formation. As shown in the following sections, increased TGF-β signaling is also critical for pathogenic changes in other related aortopathies caused by a genetic disorder.

FBN1 and TGF-β signaling pathways. Fibrillin-1 regulates the TGF-β signaling pathway by associating with LLC, consisting of LTBP and SLC. Active, free TGF-β molecules will be released by protease cleavage of inactive SLC molecules bound to LAP. LAP, latency-associated peptide; LLC, large latent complex; LTBP, latent TGF-β-binding protein; SLC, small latent complex.
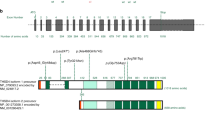
FBN1 mutations in MFS
In 1990, the locus for the Marfan mutation was mapped to chromosome 15 by linkage analysis.11 Thereafter, the first fibrillin gene mutation was found in an MFS patient in 1991, and it was confirmed that mutations in the fibrillin gene locus on chromosome 15 are responsible for MFS.12 Since then, >1500 FBN1 mutations have been identified in affected patients. Several lines of evidence have also shown that many FBN1 mutant alleles cause MFS phenotypes through a dominant-negative effect,13 though there is a considerable number of patients with mutations resulting in haplo-insufficiency owing to gene deletion, splicing mutations or nonsense mutations causing nonsense-mediated mRNA decay14 (Figure 2). Therefore, decreased protein synthesis as well as the dominant-negative effect induced by the mutant protein is thought to be a pathogenic mechanism for MFS.15 In addition, the function of fibrillin-1 has been shown to be closely related to regulation of TGF-β signaling pathways, as noted in the previous section.

NMD and haplo-insufficiency. About 30% of FBN1 mutations resulted in haplo-insufficiency while about 60% of mutations resulted in qualitative changes of fibrillin-1 molecules and about 10% of mutations resulted in splicing defect.
Loeys–Dietz syndrome (LDS) and related disorders
LDS (OMIM #609192, #610168) is an autosomal-dominant aortic aneurysm syndrome with widespread systemic involvement that shows both similarities and differences as compared with MFS.16 LDS was originally characterized by the triad of arterial tortuosity and aneurysms, hypertelorism and bifid uvula or cleft palate, though a wide range of variable phenotypes associated with this disorder have since been recognized.17 The syndrome is caused by mutations in the TGF-β receptor 1 and 2 genes (TGFBR1, TGFBR2), and diagnosis is confirmed by genotyping. Some patients have craniofacial involvement consisting of cleft palate, craniosynostosis or hypertelorism, though those do not appear in all. Bifid uvula may also be present in some but not all patients. Some reports have indicated that the natural history is characterized by aggressive arterial aneurysms while some patients show milder phenotypes.
Mutations in the TGFBR2 gene in patients with the type 2 variant of MFS and without ocular involvement have been reported.18 Most LDS patients develop aortic root aneurysms while a previous study found that the mean age at death was 26 years (range 0.5–47 years) and caused by such factors as thoracic aortic dissection, abdominal aortic dissection and intracerebral bleeding.16 Therefore, it is recognized that LDS patients tend to experience more aggressive vascular events. However, it is also known that LDS has a large variability of phenotypes including vascular lesions, as some affected patients show severe and rapid aortic events with typical craniofacial features, whereas others show mild aortic lesions without craniofacial or skeletal features.
LDS and related disorders: TGF-β signalopathies
Most LDS patients demonstrate missense mutations in the serine/threonine kinase domain of the TGF-β receptors, suggesting loss of function as the pathogenic mechanism (Figure 3). Also, there have been several reports of dysregulation of TGF-β signaling. In histochemical studies, increased TGF-β and mitogen-activated protein kinase signaling in aortic lesions of affected patients as well as in Tgfbr2 knockout mice has been found.19

TGF-β signalopathies. Several genes (FBN1, TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD3 and SKI) associated with Marfan and related disorders are indeed closely connected with the TGF-β signaling pathways. Changes in the signal regulatory function promote pathological progress of vessels.
Recently, three additional genes were identified as responsible for LDS-like phenotypes; SMAD3, TGFB2, and TGFB3. SMAD3 mutations were initially described in relation to aneurysm-osteoarthritis syndrome,20 though some patients with an SMAD3 mutation do not show prominent osteoarthritis. TGFB2 mutations have also been described in patients with mild systemic features of MFS as well as those with LDS-like features.21, 22 Very recently, TGFB3 mutations were reported in patients with syndromic types of thoracic aortic aneurysms similar to those seen in MFS and LDS,23 though one patient with a de novo TGFB3 mutation showed neither MFS nor LDS features and had no evidence of vascular disease.24
In most of these gene mutations, immunohistochemical staining reveals an increase of phosphorylated SMAD2 in aortic wall tissues, indicating that TGF-β signaling has been changed to increase the downstream gene. Therefore, in addition to the receptors, intracellular signaling molecules as well as their ligands are now considered to be responsible for the common pathogenic changes towards MFS- or LDS-related phenotypes. Nevertheless, it remains unknown how dysfunction or haploinsufficiency of these molecules results in increased TGF-β signaling even if such a change causes loss of function. Although the precise mechanisms involved in this change are uncertain, it is considered that negative feedback and non-canonical stimulus may be related to the increase in TGF-β signaling. Indeed, previous results have shown that haplo-insufficient Tgfbr2 mice as well as cranial neural crest cell-specific Tgfbr2-deficient mice recapitulate human phenotypes, such as aortic dilatations or craniofacial deformities, along with increased non-canonical (phosphorylated extracellular signal-regulated kinase) TGF-β signaling pathways.19, 25
Shprintzen–Goldberg syndrome (SGS: OMIM 182212), which results in craniosynostosis, skeletal changes (arachnodactyly, camptodactyly, scoliosis, joint hypermobility) and aortic aneurysms, shows considerable phenotypic overlap with MFS and LDS, and affected patients have been found to have mutations in the v-ski avian sarcoma viral oncogene homolog SKI.26 The oncogene SKI encodes a protein that has an important role in the negative feedback loop of the TGF-β signaling pathway. Therefore, not only signaling molecules but also molecules affecting the TGF-β signaling pathway, as well as possibly others, may be additional key molecules related to MFS- or LDS-like phenotypes, including aortopathy.
Autosomal-recessive cutis laxa type 1B and arterial tortuosity syndrome, two other rare recessive connective tissue disorders, have been shown to have autosomal-recessive gene mutations in EFEMP2,27 which codes FBLN4 proteins. FBLN4 binds tightly to LTBP1 and regulates the latency of TGF-β cytokine. Indeed, upregulated TGF-β signaling has been found in both fibroblasts of patients with FBLN4 mutations and the aortic walls of Fbln-4 hypomorphic mice.27, 28
Vascular Ehlers–Danlos syndrome (vEDS)
The vascular type of Ehlers–Danlos syndrome (type 4 EDS: OMIM #130050) caused by a deficiency of type III collagen (COL3A1),29 is a disorder featuring cutaneous, skeletal, and vascular abnormalities, including vascular rupture and easy bruising, though affected individuals usually do not show skin hyperextensibility. Clinical phenotypes include rupture of middle-sized arteries, the bowels or the uterus. Therefore, general care and follow-up examinations are critical for management of patients with vEDS based on the diagnosis, and determination of the COL3A1 mutation is critical for affected individuals and their family members. There is no evidence that COL3A1 mutations change TGF-β pathway regulation while it has been reported that celiprolol can reduce the risk for vascular events in patients with vEDS.30
It has also been reported that EDS-like phenotypes are associated with FLNA mutations.31 The FLNA gene encodes filamin A, an intermediate filament connecting the contractile apparatus of vascular smooth muscle cells to cell membranes.32 Also, filamin A has numerous interaction partners, including membrane receptors, signaling proteins and transcription factors. It is well known that FLNA mutations are associated with periventricular nodular heterotopias (OMIM #300049), though patients with those mutations may exhibit aortic root dilatation, mitral valve disease and joint hypermobility even without demonstrating periventricular nodular heterotopias.
Other types of syndromic TAAD
In addition to syndromic TAAD described above, aortic root aneurysms have also been found in other rare genetic syndromes. Sometimes, patients was not recognized to have syndromic features as key symptoms but showed TAAD events as the main clinical features. In such cases, other signaling pathways besides the TGF-β pathway seem to be involved. Aortic aneurysms have been described to sporadically occur in association with mutations involving the rat sarcoma–extracellular signal-regulated kinase signaling pathway, including Noonan syndrome pathogenic genes such as PTPN1133 and the neurofibromatosis gene, NF1.34 The fact that these genes are associated with TAAD indicates that the rat sarcoma and non-canonical TGF-β signaling pathways are involved in aortic aneurysms, as shown in MFS and LDS.
Also, the Notch signaling pathway, which is involved in Alagille syndrome,35 and genes encoding collagens or enzymes involved in collagen maturation (COL1A1/A2, COL4A1, COL4A3/4/5, PLOD3) have been shown to be associated with aortic aneurysm development. In addition, aortic aneurysm has been implicated to have an association with autosomal-dominant polycystic kidney disease,36 and aortic aneurysms and dissections of the thoracic aorta occur more frequently in patients with autosomal-dominant polycystic kidney disease.
Non-syndromic TAAD
In addition to the genes involved in syndromic types of TAAD, several others have been identified in non-syndromic familial TAAD (FTAAD) while ACTA2 mutations have been found to be responsible for approximately 15% of patients with FTAAD.37 The ACTA2 gene encodes a smooth muscle-specific isoform of the α-actin protein and is involved in contractile function of vascular smooth muscles. Patients with an ACTA2 mutation also show other vascular manifestations, such as coronary artery disease and stroke, as well as several functional disorders of smooth muscles, including vasculopathy, congenital mydriasis, patent ductus arteriosus and thoracic aortic aneurysm. In addition, some specific ACTA2 missense mutations (p.Arg179His, p.Arg258His, p.Arg258Cys) have been reported to be associated with a moyamoya-like cerebral atrial disorder.38, 39 Livedo reticularis and iris flocculi were also found in some patients with the p.Arg149Cys ACTA2 missense mutation.37
Mutations in MYH11 have also been found in patients with FTAAD and patent ductus arteriosus.40 MYH11 encodes a myosin heavy chain involved in the contractile function of vascular smooth muscles. In addition, MYLK mutations are found in a minority of FTAAD patients.41 The MYLK gene encodes myosin light chain kinase, which regulates the calcium-calmodulin-binding capacity of vascular smooth muscles by phosphorylation of myosin.
Interestingly, upregulation of TGF-β signaling in the aortic walls of TAAD patients with ACTA2 or MYH11 mutations has been reported.42 This phenomenon is related to interactions between the smooth muscle contractile apparatus and cell surface integrins that regulate TGF-β activity. However, aortic events in patients with a dysfunction of smooth muscle contractile proteins are somewhat different as compared with those in patients with a dysfunction of the TGF-β pathway, such as MFS or LDS, as FTAAD patients with an ACTA2 or MYH11 mutation demonstrate enlargement of the entire ascending aorta, which is not restricted to the sinus of Valsalva. Therefore, there may be differences regarding the pathophysiological processes involved in dysfunction of the smooth muscle contractile apparatus and those of the TGF-β pathway.
Therapy for TAAD
Following identification of the genes underlying TAAD, as noted above, clinical management stratification is based on the phenotypes related to the underlying genetic mutation. For example, LDS patients with a TGFBR1 or TGFBR2 mutation have greater risk for aortic and arterial aneurysms than other TAAD patients, and require more extensive imaging follow-up examinations to prevent vascular events. As for patients with ACTA2 mutations, the risk of coronary artery disease and stroke should be evaluated to prevent those events. The current guidelines of the American College of Cardiology43 recommend prophylactic surgery based on different scenarios according to the underlying gene as the different clinical courses are expected in patients with different gene mutations.
Medical management strategy for TAAD patients aims to reduce aortic wall stress, generally by β-blocker treatment. In addition, calcium channel blockers and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers may be given. In a study of MFS and related disorders, the discovery of the key role of TGF-β signaling in the pathogenesis of MFS mice prompted investigation of the effects of TGF-β-neutralizing treatment by a TGF-β-neutralizing antibody and losartan, an angiotensin II type I receptor antagonist, in a mouse model.44 Results showed effects of losartan on aortic root dilatation and elastic fiber fragmentation by reduction of TGF-β signaling.44 As losartan is an established drug for treatment of hypertension, investigations of its usefulness in MFS patients have begun. An initial preliminary study showed improved effectiveness for reducing progressive aortic root dilatation by losartan added to β-blocker therapy as compared with β-blocker therapy alone.45 However, the largest clinical trial to date to assess the efficacy of losartan as compared with a β-blocker found no better outcome in MFS patients who received losartan therapy.46 There are several lines of evidence showing involvement of the TGF-β pathway in MFS, LDS and related disorders. Therefore, treatments aimed to block non-canonical components of the TGF-β signaling pathway are anticipated. In addition, as changes in the vascular smooth muscle cell contractile apparatus or matrix metalloproteinase proteins are thought to have other important pathogenic roles in genetic aortic diseases, those changes will receive focus as alternative therapeutic targets. Further delineation of the genes and pathways responsible for remaining aortopathies with unknown mechanisms is also needed.
Conclusion
Since the discovery of FBN1 and COL3A1 mutations in patients with MFS or vEDS in the late twentieth century, several genes responsible for genetic aortic diseases have been identified and understanding of the related pathophysiological mechanisms has progressed. Nevertheless, progress for developing new strategies to cure these diseases remains rather slow, in spite of the rapidly increasing understanding of the physiological roles of several genes involved in these aortic diseases. Additional studies are needed for development of novel therapeutic strategies for these aortic diseases.
References
Lindsay, M. E. & Dietz, H. C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473, 308–316 (2011).
Ramanath, V. S., Oh, J. K., Sundt, T. M. 3rd & Eagle, K. A. Acute aortic syndromes and thoracic aortic aneurysm. Mayo Clin. Proc. 84, 465–481 (2009).
Blanchard, J. F., Armenian, H. K. & Friesen, P. P. Risk factors for abdominal aortic aneurysm: results of a case-control study. Am. J. Epidemiol. 151, 575–583 (2000).
Marfan, A. B. Un cas de deformation congenitale des quatre membres, plus prononcee aux extremites, caracterisee par l'allongement des os avec un certain degre d'amincissement. Bull. Mem. Soc. Med. Hop. Paris 13, 220–226 (1896).
Pyeritz, R. E. & McKusick, V. A. The Marfan syndrome. N. Engl. J. Med. 300, 772–777 (1979).
Beighton, P., De Paepe, A., Danks, D., Finidori, G., Gedde-Dahl, T., Goodman, R. et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am. J. Med. Genet. 29, 581–594 (1988).
De Paepe, A., Devereux, R. B., Dietz, H. C., Hennekam, R. C. M. & Pyeritz, R. E. Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 62, 417–426 (1996).
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B. et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet 47, 476–485 (2010).
Annes, J. P., Munger, J. S. & Rifkin, D. B. Making sense of latent TGFβ activation. J. Cell Sci. 116, 217–224 (2003).
ten Dijke, P. & Arthur, H. M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 8, 857–869 (2007).
Kainulainen, K., Steinmann, B., Collins, F., Dietz, H. C., Francomano, C. A., Child, A. et al. Marfan syndrome: no evidence for heterogeneity in different populations, and more precise mapping of the gene. Am. J. Hum. Genet. 49, 662–667 (1991).
Dietz, H. C., Cutting, G. R., Pyeritz, R. E., Maslen, C. L., Sakai, L. Y., Corson, G. M. et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991).
Aoyama, T., Francke, U., Gasner, C. & Furthmayr, H. Fibrillin abnormalities and prognosis in Marfan syndrome and related disorders. Am. J. Med. Genet. 58, 169–176 (1995).
Faivre, L., Collod-Beroud, G., Loeys, B. L., Child, A., Binquet, C., Gautier, E. et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am. J. Hum. Genet. 81, 454–466 (2007).
Judge, D. P., Biery, N. J., Keene, D. R., Geubtner, J., Myers, L., Huso, D. L. et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Invest. 114, 172–181 (2004).
Loeys, BL, Schwarze, U, Holm, T, Callewaert, BL, Thomas, GH, Pannu, H et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 355, 788–798 (2006).
Loeys, B. L., Chen, J., Neptune, E. R., Judge, D. P., Podowski, M., Holm, T. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet 37, 275–281 (2005).
Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T. et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet 36, 855–860 (2004).
Li, W., Li, Q., Jiao, Y., Qin, L., Ali, R., Zhou, J. et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest. 124, 755–767 (2014).
van de Laar, I. M., van der Linde, D., Oei, E. H., Bos, P. K., Bessems, J. H., Bierma-Zeinstra, S. M. et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 49, 47–57 (2012).
Boileau, C., Guo, D. C., Hanna, N., Regalado, E. S., Detaint, D., Gong, L. et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 44, 916–921 (2012).
Lindsay, M. E., Schepers, D., Bolar, N. A., Doyle, J. J., Gallo, E., Fert-Bober, J. et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922–927 (2012).
Bertoli-Avella, A. M., Gillis, E., Morisaki, H., Verhagen, J. M. A., de Graaf, B. M., van de Beek, G. et al. Mutations in a TGFβ ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65, 1324–1336 (2015).
Rienhoff, H. Y. Jr, Yeo, C. Y., Morissette, R., Khrebtukova, I., Melnick, J., Luo, S. et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome. Am. J. Med. Genet. 161A, 2040–2046 (2013).
Iwata, J., Hacia, J. G., Suzuki, A., Sanchez-Lara, P. A., Urata, M. & Chai, Y. Modulation of noncanonical TGF-β signaling prevents cleft palate in Tgfbr2 mutant mice. J. Clin. Invest. 122, 873–885 (2012).
Doyle, A. J., Doyle, J. J., Bessling, S. L., Maragh, S., Lindsay, M. E., Schepers, D. et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254 (2012).
Renard, M., Holm, T., Veith, R., Callewaert, B. L., Adès, L. C., Baspinar, O. et al. Altered TGFbeta signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur. J. Hum. Genet. 18, 895–901 (2010).
Huang, J., Davis, E. C., Chapman, S. L., Budatha, M., Marmorstein, L.Y, Word, R. A. et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 106, 583–592 (2010).
Superti-Furga, A., Gugler, E., Gitzelmann, R. & Steinmann, B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J. Biol. Chem. 263, 6226–6232 (1988).
Ong, K. T., Perdu, J., De Backer, J., Bozec, E., Collignon, P., Emmerich, J. et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 376, 1476–1484 (2010).
Sheen, V. L., Jansen, A., Chen, M. H., Parrini, E., Morgan, T., Ravenscroft, R. et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 64, 254–262 (2005).
Fox, J. W., Lamperti, E. D., Ekşioğlu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A. et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325 (1998).
Tartaglia, M., Mehler, E. L., Goldberg, R., Zampino, G., Brunner, H. G., Kremer, H. et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 (2001).
Wallace, M. R., Marchuk, D. A., Andersen, L. B., Letcher, R., Odeh, H. M., Saulino, A. M. et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249, 181–186 (1990).
Oda, T., Elkahloun, A. G., Pike, B. L., Okajima, K., Krantz, I. D., Genin, A. et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet 16, 235–242 (1997).
Roodvoets, A. P. Aortic aneurysms in presence of kidney disease. Lancet 1, 1413–1414 (1980).
Guo, D. C., Pannu, H., Tran-Fadulu, V., Papke, C. L., Yu, R. K., Avidan, N. et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488–1493 (2007).
Guo, D. C., Papke, C. L., Tran-Fadulu, V., Regalado, E. S., Avidan, N., Johnson, R. J. et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 84, 617–627 (2009).
Roder, C., Peters, V., Kasuya, H., Nishizawa, T., Wakita, S., Berg, D. et al. Analysis of ACTA2 in European Moyamoya disease patients. Eur. J. Paediatr. Neurol. 15, 117–122 (2011).
Zhu, L., Vranckx, R., Khau Van Kien, P., Lalande, A., Boisset, N., Mathieu, F. et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 38, 343–349 (2006).
Wang, L., Guo, D. C., Cao, J., Gong, L., Kamm, K. E., Regalado, E. et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 87, 701–707 (2010).
Renard, M., Callewaert, B., Baetens, M., Campens, L., MacDermot, K., Fryns, J. P. et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int. J. Cardiol. 165, 314–321 (2013).
Hiratzka, L. F., Bakris, G. L., Beckman, J. A., Bersin, R. M., Carr, V. F., Casey, D. E. Jr et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease: a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. J. Am. Coll. Cardiol. 55, e27–e129 (2010).
Habashi, J. P., Judge, D. P., Holm, T. M., Cohn, R. D., Loeys, B. L., Cooper, T. K. et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312, 117–121 (2006).
Brooke, B. S., Habashi, J. P., Judge, D. P., Patel, N., Loeys, B. & Dietz, H. C. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 358, 2787–2795 (2008).
Lacro, R. V., Dietz, H. C., Sleeper, L. A., Yetman, A. T., Bradley, T. J., Colan, S. D. et al. Atenolol versus losartan in children and young adults with Marfan's syndrome. N. Engl. J. Med. 371, 2061–2071 (2014).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Morisaki, T., Morisaki, H. Genetics of hereditary large vessel diseases. J Hum Genet 61, 21–26 (2016). https://doi.org/10.1038/jhg.2015.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.119
This article is cited by
-
Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene
BMC Medical Genetics (2020)
-
Congenital central pulmonary artery anomalies: Part 1
Pediatric Radiology (2020)
-
Congenital central pulmonary artery anomalies: Part 2
Pediatric Radiology (2020)
