
Abstract
Several literatures have shown the influence of geographic and linguistic factors in shaping genetic variation patterns, but their relative impact, if any, in the very heterogeneous northeastern region of Thailand has not yet been studied. This area, called Isan, is geographically structured in two wide basins, the Sakon Nakorn Basin and the Korat Basin, serving today as home to diverse ethnicities encompassing two different linguistic families, that is, the Austro-Asiatic; Suay (Kui), Mon, Chaobon (Nyahkur), So and Khmer, and the Tai-Kadai; Saek, Nyaw, Phu Tai, Kaleung and Lao Isan. In this study, we evaluated the relative role of geographic distance and barriers as well as linguistic differences as possible causes affecting the maternal genetic distances among northeastern Thai ethnicities. A 596-bp segment of the hypervariable region I mitochondrial DNA was utilized to elucidate the genetic structure and biological affinity from 433 individuals. Different statistical analyses agreed in suggesting that most ethnic groups in the Sakon Nakorn Basin are closely related. Mantel test revealed that genetic distances were highly associated to geographic (r=0.445, P<0.01) but not to linguistic (r=0.001, P>0.01) distances. Three evolutionary models were compared by Approximate Bayesian Computation. The posterior probability of the scenario, which assumed an initial population divergence possibly related to reduced gene flow among basins, was equal or higher than 0.87. All analyses exhibited concordant results supporting that geography was the most relevant factor in determining the maternal genetic structure of northeastern Thai populations.
Similar content being viewed by others


Introduction
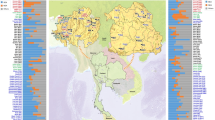
Northeastern Thailand or Isan is geographically located on the Khorat Plateau. Nearly exactly as wide as one third of Thailand, with almost the same population size, Isan shares borders with Laos to the north and the east and with Cambodia to the southeast. The Phu Phan Mountain Range straddles its northeastern interior, separating it into two wide basins, the Khorat Basin in the southwest and the Sakon Nakhon Basin in the northeast (Figure 1). Numerous archeological excavations, for example, in Ban Chiang sites, indicate that Isan was primarily inhabited by prehistoric people. The Chaobon and the Suay, who speak languages classified in the Austro-Asiatic family, sub-family Mon-Khmer, are regarded as the original inhabitants in this region before any prosperous civilizations.1 During the historical period, multiple evidence show that, prior to Angkor influence, the Isan region was dominated by two competing kingdoms: Dvaravati, a Mon-Buddhist culture from central Thailand, and Chenla, a Khmer-Hindu culture from Cambodia. During the early 9th century A.D., the Angkorian Khmer state was established. Isan was then integrated into the state as reflected by several remarkable archeological records, particularly abundant in the Khorat Basin.1, 2 In the 14th century A.D., the Khmer civilization declined and was unable to resist to the Tai-Kadai-speaking people who established the Kingdom of Lan Xang centered in Luang Prabang, in the present-day Laos. Between the late 18th and the early 19th century A.D., during the war within the Lao kingdom, the dissidents began taking refuge into the area of Isan; this represents the first documented evidence of migration from Lao to Isan region. Again in 1827 A.D., an enormous number of Lao people were forcibly migrated to Isan,3 thus increasing the dominance of Lao culture in the Isan region but, at the same time, weakening the populations of modern-day Laos.4 At that time, besides the Lao people, other ethnic groups from Laos and Vietnam migrated to the area of Northeastern Thailand, including several Tai-Kadai-speaking groups, for example, Phu Tai, Saek, Nyaw and Kaleung as well as the So, one of the Mon-Khmer-speaking populations. Most of them lived in villages along the Mekong River and its tributaries in the Sakon Nakhon Basin. In 1893 A.D., the Isan region became part of the Kingdom of Siam (Thailand) as a result of the Franco-Siamese War.1, 3

Map of Khorat Plateau showing the locations of studied populations in different geographic areas of Northeastern Thailand. Population codes are given in Table 2. Filled circles: Austro-Asiatic linguistic family; Empty symbols: Tai-Kadai linguistic family.
Isan’s long history as well as variety of ethnicities (∼18 groups populated in two distinct geographic locations) make this region an excellent area to elucidate genetic variation and its tentative influencing factors such as geography, language and culture. A general and simplifying assumption when studying linguistic variation among populations is that a common language frequently signifies a common origin and a related language indicates a common origin further back in time.5 Such linguistic relationships should be reflected by genetic variation and might be correlated with geographic distances according to a model of Isolation by Distance. Under Isolation by Distance, current patterns of genetic variation would basically result from the interaction between genetic drift (that is, random fluctuation of allele frequencies in time) and dispersal of individuals between populations, neglecting all gene flow processes other than those in which movements of individuals from their birthplaces are local and random.6, 7, 8 The correspondence between geographic, genetic and linguistic distances would hence be explained by this simple model, except in those cases in which complicating dynamics would affect the expected relationship between geographical distances and genetic and linguistic diversity. These complicating dynamics can be represented by processes of linguistic assimilation during migrations or by the presence of migration resistance factors, that is, geographical barriers to gene flow. In some cases, even language differences themselves can somehow act as a barrier to free gene flow, enhancing the genetic differentiation.9, 10, 11, 12 In Thailand, where both geographic and ethno-linguistic diversities exist, our previous researches showed the influence of both linguistics and geography on genetic diversity of peoples residing exclusively in the North of Thailand.13, 14, 15 However, it is still not clear how, and to what extent, these two factors are related with the genetic variation of Northeastern Thai populations.
Maternal inherited mitochondrial DNA (mtDNA) has been proven to be a powerful genetic marker to infer population history in regional and continental frameworks,16, 17, 18 however, until now, only four studies on genetic variation of five Northeastern Thai populations (that is, Phutai, Chaobon, Thai Khon Kaen, Thai Khorat, Thai Isan) have been published.19, 20, 21
In the present study, we analyzed new mtDNA data of 10 Isan ethnicities speaking languages belonging to two major families, namely the Tai-Kadai (Saek, Nyaw, Phu Tai, Kaleung, and Lao Isan) and the Austro-Asiatic (So, Suay, Mon, Chaobon and Khmer), and inhabiting two geographically separated wide basins, namely the Sakon Nakhon Basin (Saek, Nyaw, Phu Tai, Kaleung and So) and the Khorat Basin (Lao Isan, Suay, Mon, Chaobon and Khmer) to evaluate the relative role of geographic distance and barriers and linguistic differences as possible causes affecting the maternal genetic distances among northeastern Thai ethnicities.
Materials and methods
Samples and DNA extraction
We studied 433 maternally unrelated individuals (for at least three generations) from 10 ethnic groups, namely Khmer (KHM), Mon (MON), Suay (SUY), Chaobon (BON), So (SOA), Lao Isan (LAO), Phu Tai (PUT), Nyaw (YOH), Saek (SAK) and Kaleung (KAL), of the Northeast of Thailand. The studied populations were linguistically classified into two groups, Austro-Asiatic (AA) and Tai-Kadai (TK), and geographically separated into two groups, Sakon Nakhon (SN) Basin and Khorat (KR) Basin (Table 1 and Figure 1). General information about the studied populations are listed in Table 1. Prior to sample collection, information on linguistic, cultural aspects, village and individual history was obtained by interview and the informed consent was signed. Buccal swabs were collected from each subject by using a brush embedded in Gentra Puregene Buccal Cell Kit (Qiagen, Hilden, Germany). Genomic DNA was extracted from the collected buccal cells using Gentra Puregene Buccal Cell Kits according to the manufacturer’s protocols. The use of human subjects for this study was approved by Ethics Committee for Human Research of Khon Kaen University, Thailand.
mtDNA amplification and sequencing
The mtDNA control region (np15704-430) of the 10 ethnic groups was amplified using published primer pairs (LLmt-A, 15704- 5′-CATAGCCAATCACTTTATTG-3′-15723; LHmt-E, 430-5′-CTGTTAAAAGTGCATACCGCC-3′-410).22 PCR reactions were performed by using nPfu-Forte DNA polymerase (Enzynomics, Daejeon, Korea). Each PCR reaction mix had a final volume of 50 μl consisting of 5 μl of 10 × nPfu-Forte buffer, 5 μl of 200 μM dNTP mixture, 2.5 μl of each 5 μM PCR primer, 0.5 μl of 2.5 U μl−1 Pfu polymerase, 0.5 μl of 50 ng genomic DNA and 34 μl of distilled water. PCR reactions were performed under the following conditions: 2 min at 95 °C for an activation step, followed by 35 cycles of 30 s denaturation at 95 °C, 1 min primer annealing at 56 °C and 1 min extension at 72 °C, and 5 min at 72 °C for a final extension step. After visualization on a 1% agarose gel with a 100-bp DNA ladder (Norgen Biotek Corp., Thorold, ON, Canada), amplicons (∼1200 bp) were sent for purification and sequencing of hypervariable region I (HVRI) (np 15897-100) with a published set of primers19 (SeqLmt-A, 15897-5′-GTATAAACTAATACACCAGTCTTGT-3′-15921; SeqHmt-E, 100-5′-CAGCGTCTCGCAATGCTATCGCGTG-3′-76) at Macrogen Inc., Seoul, Korea. The sequencing results were edited, assembled and aligned with the revised Cambridge Reference Sequence23 using SeqScape software v2.7 (Applied Biosystem, Foster City, CA, USA). The HVRI sequences of all samples were submitted to GenBank (accession numbers KJ205639-KJ206068).
Statistical analyses
Genetic variation within population and demographic parameters
We identified the polymorphic sites of the mtDNA sequences of 596 nucleotides (np 16001–16569) using DnaSP v.5 software.24 Parameters of genetic diversity within populations, that is, mean pairwise differences (MPD) or intra-MPD, number of segregating sites (S), nucleotide diversity (π), number of observed haplotypes, and the haplotype diversity (h)25 were calculated by Arlequin v.3.5.26 The demographic expansion parameters, that is, a raggedness index value (r)27 as well as neutrality estimators such as Fu’s Fs28 and Tajima’s D29, were computed by using the same software. The number of shared haplotypes was determined for each of the 45 possible population pairs by a simple gene-count method.
To compare the genetic variation among populations, we calculated the MPD among populations (inter-MPD) and a measure of genetic distance between pairs of populations based on pairwise difference (Fst, significance tested by permutation). To characterize population affinity, the genetic distance matrix was then plotted in three dimensions by means of multidimensional scaling (MDS) using the available Statistica v.10 demo (StatSoft Ltd.). Spatial analysis of molecular variance (SAMOVA) in SAMOVA v.1.0 program was used to infer the most supported genetic structure of the sample, defining groups of populations that are geographically and genetically very similar.30
Genetic variance at three hierarchical subdivisions (within individuals of population, among populations within a group, and among groups of populations), was assessed by the analysis of molecular variance procedure31 as implemented in Arlequin v. 3.5. In this analysis, studied populations were grouped by both geography and language (see Table 1).
Mantel test
The correlations and partial correlations between distance matrices of genetics-geography, genetics-language, and geography-language were performed by the Mantel test.32 Table 2 shows the matrices of geographic and linguistic distance we used for the Mantel test, whereas genetic distance (Fst) matrix is shown in Table 3. Geographic distances in Km between the approximate locations of each population were computed as great-circle distances calculated from their latitudinal and longitudinal coordinates. Linguistic distances between pairs of populations were defined as simple dissimilarity indices on the basis of the hierarchical classification of languages reported in Ethnologue.33 Populations speaking languages belonging to different subfamilies, that is, AA and TK, were assigned dLAN of four, whereas different branches within subfamilies were assigned dLAN of three. Different sub-branches within branch were assigned dLAN of two and then dLAN of similar sub-branches was one.
Approximate Bayesian computation
To deeply investigate the evolutionary relationship among populations, an Approximate Bayesian Computation (ABC) procedure was applied.34 An ABC approach, which combines the analysis of large genetic data sets and realistic models, can be briefly summarized as follows: millions of genetic data sets with the same features as the observed one, that is, number of individuals, type of genetic markers, length of sequences, are generated according to the coalescent theory for each demographic model, taking into account the associated prior distributions. The pattern of genetic variation in the observed and simulated data, summarized by a certain number of statistics, is then compared by Euclidean distance. The coalescent-based simulations were performed by combinations of parameters for a specific demographic model. Those coalescent-based simulations, which generated summary statistics closest to the observed ones, as shown by smallest Euclidean distances, were then considered for calculating the posterior probabilities of each model using two different approaches, acceptance-rejection procedure (AR) and weighted multinomial logistic regression (LR).35, 36 Under the AR, the posterior probability of a model is obtained by considering only a certain number of ‘best’ simulations, and then simply counting the proportion of these retained simulations that have been generated by each model under investigation. This method can be considered reliable only when applied to a few simulations showing an excellent fit with the observed data, that is, few hundreds.36 Under LR procedure, a LR is fitted where the model is the categorical dependent variable in the ABC simulations and the summary statistics are the predictive variables. The regression is local around the vector of observed summary statistics, and the probability of each model is finally evaluated at the point corresponding to the observed vector of summary statistics. The β coefficients of the regression model are estimated by maximum likelihood and the standard errors of the estimates might be taken as a measure of the accuracy of the method. To evaluate the stability of the models’ posterior probabilities, we considered different thresholds by considering different number of retained simulations for both the model selection procedures (100, 200, 300, 500 best simulations for AR and 25 000, 50 000, 75 000, 100 000 best simulations for LR). To generate the simulated data sets, we used the software package ABCtoolbox,37 running 500 000 simulations for each model. To calculate the models’ posterior probabilities, we used R scripts from http://code.google.com/p/popabc/source/browse/#svn%2Ftrunk%2Fscripts, modified by SG. To summarize the genetic information contained in the data, we calculated the following statistics within and between populations: the number of haplotypes (h), the number of private polymorphic sites (S), Tajima’s D, intra- and inter-MPD, and pairwise Fst.
Testing the best-fit demographic models and type I error
Based on geographic locations and linguistic affiliations of the studied populations, SOA and LAO were the variable populations whose languages differ from their geographically grouped neighbors. Therefore, three demographic models were proposed to describe different aspects of the evolutionary relationships among studied populations, in which geography or language was fitted to describe mtDNA data (Figure 2). In Model 1, the separation of the lineages follows the linguistic affiliation, with a first split (Ts1) involving the AA and the TK groups, and a subsequent separation by geographic location at Ts2 (AA and SOA), and Ts3 (TK and LAO). In Model 2, the ‘driving force’ of the genetic variation is represented by geography. A first separation (Ts1) is started between populations from KR Basin and from SN Basin. Within each geographical group, the LAO and SOA, who speak different languages from their neighbors, were subsequently separated at Ts2 and Ts3, respectively. Model 3 extends Model 1, in which after Ts2 and Ts3, geographically closer populations (LAO-AA; SOA-TK) start to exchange migrants at a certain rate [m1 (m1_b) and m2 (m2_b)]. The effective population sizes were assumed to be constant in time; the prior distributions were all uniform (log-uniform for the effective population sizes), and, where possible, based on historical records (for details, see Supplementary Table 1).

Schematic presentation of the three models described by language (Model 1), geography (Model 2), and recent migration (Model 3). Ne, Ts and m are the effective population sizes, separation times and the migration rates, respectively. Population codes are given in Table 2.
We estimated the probability that the true null hypothesis would be rejected by evaluating the type I error. The proportion of cases in which 1000 pseudo-observed data sets, generated under each model, is not correctly identified by the ABC analysis (both AR and LR procedures, 100 and 50 000 retained simulations in turn). The power of the model choice procedure has been evaluated using a wide range of decision probability thresholds to identify the support for a specific model, that is, 0.5, 0.6, 0.7, 0.8, 0.9.
Results
Genetic diversity and demographic expansion
A total of 173 distinct mtDNA haplotypes were observed in 433 individuals. Among the observed haplotypes, 142 types were unique within populations, whereas the other 31 types were shared between two or more populations. Out of the 142 unique haplotypes, 34 were shared by two or more individuals within one group (multiple unique), whereas the remaining 108 haplotypes belonged to each individual (single unique). The highest number of shared haplotypes (six haplotypes) was found between SOA-PUT, but none were shared among five pairs of populations: MON-BON, MON-LAO, MON-PUT, BON-KHM and BON-SOA.
Genetic diversity within population and population expansion results are reported in Table 1. Haplotype diversity (h) varied from 0.9899 (LAO) to 0.7920 (SAK), which was in the same range as previous published populations in Thailand,13, 14, 15, 19, 20, 21 albeit rather a low h value was found in SAK, indicating possible drift effect. The lowest nucleotide diversity (π) was observed in MON (0.0098), whereas PUT had the highest value (0.0153). The intra-MPD ranged from 5.5254 (MON) to 8.6956 (PUT), reflecting genetic homogeneity or recent diverged mtDNA within the MON and genetic heterogeneity in the PUT.
The highly significant negative values of the Fu’s Fs (P<0.05) were predictions of demographic expansion in KHM, MON, SOA, LAO, and PUT. The lower raggedness index (<0.03) as well as the unimodal mismatch distribution graph for these populations (data not shown) also provide congruent evidence for population growth and expansion.38
Genetic relationships
Among 45 pairwise Fst comparisons, 36 (80%) were statistically significant (P<0.01) (Table 3). The MON, BON, and SAK showed significant Fst values for all comparisons, indicated high genetic differentiation. The KHM had genetically differentiated from almost all other populations, except the LAO. It is interesting that SUY and BON have genetic similarity. Most Fst comparisons between populations in SN Basin were not statistically significant, particularly among the SOA, PUT and YOH, reflecting genetic homogeneity. The corrected MPD among populations showed a similar pattern to Fst result (see Supplementary Table 2), which indicates that the SAK were most differentiated, whereas the next most respectively differentiated populations were the BON, MON and SUY.
To visualize the genetic relationship among populations, we plotted a pairwise Fst matrix through MDS analysis and performed SAMOVA analysis. In the MDS as shown in Figure 3, most populations residing in the SN basin (YOH, PUT, SOA and KAL) were clustered in the center of the plot with the exception of the SAK, which appear to be the most genetically differentiated population, even if still genetically more closely related to neighbors in the SN Basin than to populations from the KR basin. Surprisingly, although the LAO resided in the KR Basin, they clustered together with other SN dwelling populations. For the ethnicities located in the KR basin, the KHM were quite genetically proximate to the SUY and BON, whereas the MON was considerably distanced from other studied populations, indicating their genetic distinction. In SAMOVA analysis, when number of group was increasing from two-groups until six-groups category, the SAK, MON, SUY, BON and KHM, respectively, were partitioned from the other populations (Table 4). The maximal percent of variation with significant value was observed at six-groups category (7.287%, P<0.01): SAK, MON, SUY, BON, KHM, YOH-PUT-SOA-KAL-LAO. Interestingly, population grouping by SAMOVA was concordant to MDS plot.

Three dimentional scaling plot (3D-MDS) constructed based on pairwise Fst. Population codes are given in Table 1. Stress value for MDS=0.0339.
Correlation among genetics, geography and language
The analysis of molecular variance was used to infer the proportion of total genetic variation accounted by groups. Groupings were defined on the basis of geographic and linguistic classification (Table 5). When populations were grouped according to geography, the results revealed that it can be used to describe the genetic structure of studied populations, as the amount of observed variation among groups was 4.68% with statistical difference (Fct=0.0468, P<0.01), whereas the proportion of variance among population within groups explain 7.429% (Fsc=0.07429, P<0.01) and within populations explain 11.765% (Fst=0.11765, P<0.01). The average Fst of populations in the SN basin (Fst=0.06902, P<0.01) and in the KR basin (Fst=0.07900, P<0.01) were much lower than the overall Fst (0.09889, P<0.01). It seems evident that there is a certain level of genetic homogeneity among populations within each geographic region, with an higher homogeneity in populations from the SN basin than in populations from the KR basin.
Based on linguistic classification, the proportion of genetic variation among groups was considerably low (0.913%) with no statistical significance (Fct=0.00913, P>0.01), reflecting no relationship between genetic distance and linguistic affiliation. Most of the genetic variance (89.74%) was found within populations (Fsc=0.09434, P<0.01), whereas variance among populations within the linguistic groups was 9.35% (Fst=0.10260, P<0.01). We observed a slight higher value of average Fst of AA (Fst=0.10681, P<0.01) with respect to Tai speaking group (Fst=0.07820, P<0.01), possibly indicating more genetic heterogeneity among AA than among TK groups. A notable amount of genetic variance was found among geographic groups, which is higher than variance among linguistic groups.
Mantel testing showed that genetic distances strongly correlated to geographic distances by means of correlation test (r=0.4713, P<0.01) and partial correlation test (r=0.4449, P<0.01), whereas, we detected no correlation and partial correlation between genetic and linguistic distances (r=0.1735, P>0.01 and r=0.0008, P>0.01, respectively). Among geographic and linguistic matrices, no correlation (r=0.3667, P>0.01) and partial correlation (r=0.3281, P>0.01) was observed.
Model selection
Table 6 shows the posterior probabilities of the three considered evolutionary scenarios. Model 2, in which the geography has a major role in shaping the genetic variation, received the strongest support. The posterior probability of Model 2 was never lower than 87%, considering both AR and LR and remained stable over different number of retained simulations. To assess the reliability of the probabilities estimated, we also evaluated the models’ posterior probabilities within two times the range of the standard error associated to the β coefficients of the regression model (in both directions). The support remained in favor of Model 2.
To evaluate whether there is enough power in the data for these models to be discriminated, we calculated a type I error, that is, the incorrect rejection of a true null hypothesis. This analysis has fundamentally verified the reliability of the estimated probabilities because the compared models were quite similar to each other, and only a single genetic locus was analyzed. The type I error analysis considered both AR and LR as criterion of model selection, and several probability thresholds to identify the support for a specific model (Table 7). The results of the LR (50 000 best simulations) were in general better than those obtained with the acceptance-rejection, especially for higher probability thresholds. The models appeared to be well recognized even when the decision probability threshold was 0.5, as the probability of recognizing the right model was never lower than 0.6. Moreover, when the right model was not selected as the ‘true’ one, the alternative models almost never reached a probability high enough to be supported by the ABC model selection procedure. This was particularly true when the decision probability threshold was very high (0.9), that is, similar to the value we obtained from the real data for Model 2. All together these results can be considered highly significant, and the model that has been selected here (Model 2) can be confidently regarded as the best one.
Discussion
MtDNA sequences data analyzed in the current study provide us a better understanding about the level of genetic variation in a micro-geographic scale and about past population dynamics in several ethnicities of the Isan region or Northeastern Thailand. Whether geography or language most influenced genetic variation of populations within regional, continental, and worldwide scales have been long-standing questions for molecular anthropologists and human geneticists.39, 40, 41, 42, 43 To date, there has been no report aimed to answer the above question for populations residing in the Northeast of Thailand, addressed here for the first time. Results obtained from Mantel test, AMOVA, SAMOVA and ABC procedures indicate that geography has an important role to determine northeastern Thai genetic structure, according to Isolation by Distance model. Under Isolation by Distance, current patterns of genetic variation would simply result from the interaction between genetic drift and dispersal of individuals between populations, thus resulting in a decrease of genetic similarities between populations when geographic distance increases.6 Based on linguistic and archeological data, each of the two different geographic regions in Isan was occupied by linguistically distinct groups of people. The native AA populations were mainly resided in KR basin,44 whereas the TK people who migrated from Laos and Vietnam were centered in the SN Basin.3
Living in close geographical proximity, the SOA and their neighbors (PUT, SAK, KAL and YOH), have languages of unrelated ancestry. Our genetic findings reveal the parallelism between genetic variation and geographic factors. The SOA’s historical homeland is in the forest-covered hills of Khammuan Province in Laos. Some of the tribe members migrated to Thai soil, in the area of Kusumal District, in 1844 A.D.1 Based on ABC procedure (Figure 2), the three demographic scenarios might be suggested to explain the degree of genetic resemblance between SOA and other SN populations, possibly linked to a recent common origin. The SOA and other neighbors within SN Basin might have shared genetic similarity from the time they resided in their historical homeland in Laos and Vietnam. At that time, they might have come into contact and after the migratory time with spatial and temporal different settlement in Thailand, their genetic homogeneity continued. The two greatest numbers of shared haplotypes between SOA-PUT and SOA-YOH, respectively, could be additionally explained by the same genetic source between the SOA and their neighbors.
In accordance with pairwise Fst, MPD and SAMOVA, the peculiar genetic divergence of the SAK made this population particularly interesting. Historically, the SAK originated in Vietnam and then with the influence of the Kinh (the vast majority of Vietnamese), they moved westward to Laos around 380 years ago. The majority of SAK are centered in Khammuan Province of Laos and they migrated across the Mekong river into Nakorn Panom Province of Thailand about 200 years ago.1 The greatest differentiation as seen in the SAK is likely a consequence of genetic drift associated with female immigrants during the settlement period. The limited genetic diversity, as reflected by the lowest haplotype diversity and second lowest intra-MPD (Table 1), were regarded as reliable indicators of a genetic bottleneck.45 The debates on the origin of the SAK have arisen in linguistic classification. At first, the language of the SAK was classified as belonging to the AA family in the Mon-Khmer sub-family, but later most linguists classified the SAK language to the TK family in the Northern Tai branch, spoken mainly by the Tai in Gwangsi Province of China.1, 46 The SAK exhibited closest genetic relationship to the SOA. It might be indicated that the SAK are genetically more closely related to AA than to TK groups. Thus, based on several articles reporting the strong association between linguistic and genetic classifications,47, 48, 49, 50 to our knowledge, the present-day SAK language classification is not in agreement with genetic affinity. However, it should be cautioned that the genetic ancestry of the SAK might be blurred by strong influences of the geographic factor.
Almost all AA groups, KHM, BON, SUY and MON, as well as the only TK village of the LAO, were dispersedly situated in the KR basin. LAO or Lao Isan refers to peoples who are ethnically Lao but are Thai citizens.4 They comprise the majority of inhabitants and are widely distributed in all provinces of Northeastern Thailand. Most of Lao Isan people were forcibly migrated from their historical homeland in the present-day Laos during 1827–1870 A.D.3 Although the LAO village in this study was located within the area of KR Basin, close genetic affinity between the LAO and populations in the SN Basin was detected. Through previous massive migration, the LAO in the SN and KR Basins might have still preserved genetic similarity, thus, close genetic relationship might have resulted in low levels of differentiation between LAO populations in the SN and KR Basins. Future study with more broad samples of LAO from the SN Basin will be helpful to evaluate this assumption.
Interestingly, nonsignificant pairwise Fst between LAO and KHM could be plausibly explained by extensive gene flow, concordant with an earlier genetic study,20 and socio-linguistic research.51, 52, 53 Although current study’s results support that geography explains genetic variation and relationship among populations, we somehow detect significant genetic differentiations among populations within the KR Basin. It might be suggested that geographic proximity determined the genetic homogeneity among AA populations in the past, but later on, the factors of cultural and linguistic differences as well as evolutionary factors, like drift effect, inbreeding and genetic exchange, overcame the influence of spatial isolation, as reflected in KHM, BON, SUY and MON.
A certain degree of inbreeding is evident particularly in the Chaobon (BON), alternately called Nyahkur. Chaobon inhabited the area that is now Thailand preceding the coming of the Khmer and the Tai groups. They now lived in Thailand only in Chaiyabhum, Petchaboon and Nakorn Rachasima provinces (Figure 1). The bulk of these people live in Chaiyabhum Province, scattered among different deep jungle and mountainous villages.1, 54, 55 The most original Chaobon tribe in Wang Ai Pho village in Chaiyabhum Province, who still preserved their language and culture, was sampled in this study. Loss of genetic diversity, as indicated by low values of h, S and intra-MPD, might reflect consanguineous marriage due to cultural isolation. This study has documented the sequential genetic effects from preserved cultural practice within this population before they may be possibly erased by the opportunity for admixture with Lao Isan people. Based on linguistic research, Chaobon are believed to be the remaining descendants of the ancient Mon from the historic Dvaravati period. Contrary to our expectation, the present results do not support the genetic bond between the extant BON and MON.
The Mon are one of the oldest settlers in Southeast Asia. Their origin is uncertain. It is known that they once lived in Southwest China, and moved down to upper Myanmar early in the Christian era. They were politically driven southward to settle in Pegu and Thaton, in Myanmar and eastward to the present-day Central and Southern Thailand, respectively. The great Mon Dvaravati Kingdom with an advanced civilization was founded between the 3rd and 10th century A.D. in the area of Central Thailand.1 The prosperous Mon Kingdom expanded to present-day Southern, Northern and Northeastern Thailand. In 1775 A.D., the first group of studied Mon migrated from Myanmar to settle down in Nakorn Rachasima, further increasing in population size to ∼2500 around 1793 A.D. The studied MON who historically migrated from Myanmar was indeed different from Dvaravati Mon in Central Thailand, therefore a genetic link between BON and MON was not apparent. Another important finding emerged from the results of genetic diversity and demographic expansion parameters, which exhibit the lowest π, intra-MPD and number of multiple unique haplotypes. These, as well as positive signals of population growth in the MON (Table 2), provide congruent evidence for a recent bottleneck followed by an expansion in the population, which have not yet been recognized in socio-linguistic and historic literatures.
Worthy of attention is the genetic ancestry of the Suay (SUY). MDS result reveals the close genetic relatedness between SUY and KHM, whereas pairwise Fst indicates nonsignificant genetic difference between SUY and BON. These results seem to be congruent with previous historic research documenting connections between SUY and KHM in language, history, society and ancestry. The Suay or Kui, called Kamen-boran (meaning ancient Khmer) by Khmer people, are the original inhabitants of part of Thailand, Laos and Cambodia, predating the invasion of the Khmer and the Tai group. Nowadays Suay in Thailand have been adopted a Thai-Lao language referred to as Lao-Suay or a Khmer language referred to as Khmer-Suay.1 The current studied Suay from Surin Province migrated at first from Southern Laos during 1656–1688 A.D. and then sporadically moved until around 1760 A.D. when the mass migration period occurred.56 However, it has been proposed by some scholars14 that SUY share ancestry with BON, now strengthened by our investigation.
To summarize, this study highlighted some main aspects of maternal genetic structure of various populations in Northeastern Thailand. Genetic findings obtained through this study made it possible to infer the influence of geographic factors in shaping patterns of genetic variations and affinity among linguistically diverse populations. Genetic divergence between populations was primarily influenced by geography. Then, within the same geographic location different driving forces, including language and culture as well as evolutionary driven factors, like genetic drift from founder effect, inbreeding and admixture are considered to be the plausible additional factors. Our results remain open to future investigations with further mtDNA sequences from other populations and genetic data from different genetic markers to gain more insight into genetic history of Northeastern Thai people.
References
Schliesinger, J. Ethnic groups of Thailand: Non-Tai-speaking peoples, (White Lotus Press: Bangkok, Thailand, 2000).
Wongtaed, S. Explore Isan Society and Cultures, (Art and culture Press: Bangkok, Thailand, 1999).
Schliesinger, J. Tai Group of Thailand, Vol 1: Introduction and overview, (White Lotus Press: Bangkok, Thailand, 2001).
Bonnie Pacala, B. & Somroay, Y. Buddhist murals of northeast Thailand: Reflection of the Isan heartland, (Silkworm Books: Chiangmai, Thailand, 2010).
Ruhlen, M. A Guide to the World’s Languages Vol. 1 Classification (Stanford University Press: Stanford, CA, 1987).
Wright, S. Isolation by distance. Genetics 28, 114–138 (1943).
Slatkin, M. Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47, 264–279 (1993).
Barbujani, G. Geographic patterns: how to identify them and why. Hum. Biol. 72, 133–153 (2000).
Sokal, R. R. Genetic, geographic and linguistic distances in Europe. Proc. Natl Acad. Sci. USA 85, 1722–1726 (1988).
Zerjal, T., Beckman, L., Beckman, G., Mikelsaar, A. V., Krumina, A., Kucinskas, V. et al. Geographical, Linguistic and Cultural Influences on Genetic Diversity: Y-Chromosal Distribution in Northern European Populations. Mol. Biol. Evol. 8, 1077–1087 (2001).
Cavalli-Sforza, L. L., Minch, E. & Mountain, J. L. Coevolution of genes and languages revisited. Proc. Natl Acad. Sci. USA 89, 5620–5624 (1992).
Chaubey, G., Metspalu, M., Karmin, M., Thangaraj, K., Rootsi, S., Parik, J. et al. Language shift by indigenous population: A model genetic study in South Asia. Int. J. Hum. Genet. 8, 41–50 (2008).
Besaggio, D., Fuselli, S., Srikummool, M., Kampuansai, J., Castrì, L., Tyler-Smith, C. et al. Genetic variation in Northern Thailand Hill Tribes: origins and relationships with social structure and linguistic differences. BMC Evol. Biol. 7 (Suppl 2), S12 (2007).
Kutanan, W., Kampuansai, J., Nakbunlung, S., Lertvicha, P., Seielstad, M., Bertorelle, G. et al. Genetic structure of KhonMueang populations along a historical Yuan migration route in Northern Thailand. Chiang Mai J. Sci. 38, 295–305 (2011a).
Kutanan, W., Kampuansai, J., Fuselli, S., Nakbunlung, S., Seielstad, M., Bertorelle, G. et al. Genetic structure of the Mon-Khmer speaking groups and their affinity to the neighbouring Tai populations in Northern Thailand. BMC Genet. 12, 56 (2011b).
Cavalli-Sforza, L. L. & Feldman, M. W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 33 (Suppl), 266–275 (2003).
Alfonso-Sánchez, M. A., Cardoso, S., Martínez-Bouzas, C., Peña, J. A., Herrera, R. J., Castro, A. et al. Mitochondrial DNA haplogroup diversity in Basques: a reassessment based on HVI and HVII polymorphisms. Am. J. Hum. Biol. 20, 154–164 (2008).
Malyarchuk, B. A., Perkova, M. A., Derenko, M. V., Vanecek, T., Lazur, J. & Gomolcak, P. Mitochondrial DNA variability in Slovaks, with application to the Roma origin. Ann. Hum. Genet. 72, 228–240 (2008).
Fucharoen, G., Fucharoen, S. & Horai, S. Mitochondrial DNA polymorphisms in Thailand. J. Hum. Genet. 46, 115–125 (2001).
Lertrit, P., Poolsuwan, S., Thosarat, R., Sanpachudayan, T., Boonyarit, H., Chinpaisal, C. et al. Genetic history of Southeast Asian populations as revealed by ancient and modern human mitochondrial DNA analysis. Am. J. Phys. Anthropol. 137, 425–440 (2008).
Kutanan, W., Srithawong, S., Kamlao, A. & Kampuansai, J. Mitochondrial DNA-HVR1 variation reveals genetic heterogeneity in Thai-Isan peoples from the lower region of Northeastern Thailand. Adv. Anthropol. 4, 7–12 (2014).
Schurr, T. G., Sukernik, R. I., Starikovskaya, Y. B. & Wallace, D. C. Mitochondrial DNA variation in Koryaks and Itel’men: population replacement in the Okhotsk sea-Bering sea region during the Neolithic. Am. J. Phys. Anthropol. 108, 1–39 (1999).
Andrews, R. M., Kubacka, I., Chinnery, P. F., Lightowlers, R. N., Turnbull, D. M. & Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 (1999).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Nei, M. Molecular Evolutionary Genetics, (Columbia University Press: New York, USA, 1987).
Excoffier, L. & Lischer, H. E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Eco. Res. 10, 564–567 (2010).
Harpending, H. C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66, 591–600 (1994).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Dupanloup, I., Schneider, S. & Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 11, 2571–2581 (2002).
Excoffier, L., Smouse, P. & Wuattro, J. Analysis of molecular variance inferred from metric distance among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
Lewis, M. P. Ethnologue: Languages of the World 16th edn (SIL International: Dallas, TX, USA, 2009) Online version http://www.ethnologue.com/.
Bertorelle, G., Benazzo, A. & Mona, S. ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Mol. Ecol. 19, 2609–2625 (2010).
Pritchard, J. K., Seielstad, M. T., Perez-Lezaun, A. & Feldman, M. W. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16, 1791–1798 (1999).
Beaumont, M. Joint determination of topology, divergence time and immigration in Simulation, Genetics, and Human Prehistory eds Matsumura S., Forster P., Renfrew C., 135–154 (McDonald Institute for Archaeological Research: Cambridge, England, 2008).
Wegmann, D., Leuenberger, C., Neuenschwander, S. & Excoffier, L. ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC Bioinformatics 11, 116 (2010).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9, 552–569 (1992).
Helgason, A., Yngvadóttir, B., Hrafnkelsson, B., Gulcher, J. & Stefánsson, K. An Icelandic example of the impact of population structure on association studies. Nat. Genet. 37, 90–95 (2004).
Pardiñas, A. F., Roca, A., García-Vazquez, E. & López, B. Assessing the Genetic Influence of Ancient Sociopolitical Structure: Micro-differentiation Patterns in the Population of Asturias (Northern Spain). PLoS ONE 7, e50206 (2012).
Cavalli-Sforza, L. L., Menozzi, P. & Piazza, A. The history and geography of human genes, (Princeton University Press: Princeton, USA, 1994).
Eller, E. Population substructure and isolation by distance in three continent regions. Am. J. Phys. Anthropol. 108, 147–159 (1999).
Coia, V., Boschi, I., Trombetta, F., Cavulli, F., Montinaro, F., Destro-Bisol, G. et al. Evidence of high genetic variation among linguistically diverse populations on a micro-geographic scale: a case study of the Italian Alps. J. Hum. Genet. 57, 254–260 (2012).
Premsrirat, S. Linguistic contributions to the study of the Northern Khmer language of Thailand in the last two decades. Mon-Khmer Stud. 27, 129–136 (1997).
Davis, M. C., Novak, S. J. & Hampikian, G. Mitochondrial DNA analysis of an immigrant Basque population: loss of diversity due to founder effects. Am. J. Phys.Anthropol. 144, 516–525 (2011).
Smalley, W. A. Linguistic Diversity and National Unity: Language Ecology in Thailand, (University of Chicago Press: Chicago, USA, 1994).
Barbujani, G. & Sokal, R. R. Zones of sharp genetic change in Europe are also linguistic boundaries. Proc. Natl Acad. Sci. USA 87, 1816–1819 (1990).
Cavalli-Sforza, L. L., Minch, E. & Mountain, J. L. Coevolution of genes and languages revisited. Proc. Natl Acad. Sci. USA 89, 5620–5624 (1992).
Barbujani, G. & Pilastro, A. Genetic evidence on origin and dispersal of human populations speaking languages of the Nostratic Macro family. Proc. Natl Acad. Sci. USA 90, 4670–4673 (1993).
Boattini, A, Griso, C. & Pettener, D. Linguistic versus genetic isolation.The strange case of the Walser from Upper Lys Valley (Italian Western Alps). J. Anthropol. Sci. 89, 161–175 (2011).
Smalley, W. A. Multilingualism in the Northern Khmer population of Thailand. Language Sci. 10, 395–408 (1988).
Khanittanan, W. Khmero-Thai: the great change in the history of Thai Language in the Chao Praya basin. J. Lang Linguist. 19, 35–50 (2001).
Talbot, S. & Janthed, C. Northeast Thailand before Angkor: Evidence from an Archaeological Excavation at the Prasat Hin Phimai. Asia Perspective 40, 179–194 (2002).
Premsrirat, S. The Future of NyahKur in Collected papers on Southeast Asian and Pacific languages eds Bauer R. S., 155–165 (The Australian University: Canberra, Australia, 2002).
Prasert, S., Pansila, V. & Lasunon, O. Guidelines and Methods for Conservation, Revitalization and Development of the Traditions and Customs of NyahKur Ethnic Group for Tourism in the Province of Chaiyapum in Northeast Thailand. The Social Sciences 4, 174–179 (2009).
Sa-ard, O. Phrase to sentence in Kuay (Surin), (Mahidol University: Nakorn Pathom, Thailand, 1984).
Premsrirat, S. Languge map of ethnic groups in Thailand. J. Lang. Cult. 21, 5–35 (2002) in Thai.
Acknowledgements
We would like to thank village chiefs and all voluntary donors. We also thank Dr Alvin Yoshinaga for English approval on this manuscript. WK was supported by Thailand Research Fund (TRF) (Grant No. MRG5580058). SF was funded by European Research Council Advanced Grant Agreement 295733, ‘LanGeLin’ Project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kutanan, W., Ghirotto, S., Bertorelle, G. et al. Geography has more influence than language on maternal genetic structure of various northeastern Thai ethnicities. J Hum Genet 59, 512–520 (2014). https://doi.org/10.1038/jhg.2014.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.64
This article is cited by
-
Forensic and genetic characterizations of diverse southern Thai populations based on 15 autosomal STRs
Scientific Reports (2022)
-
Complete mitochondrial genomes of Thai and Lao populations indicate an ancient origin of Austroasiatic groups and demic diffusion in the spread of Tai–Kadai languages
Human Genetics (2017)
-
Genetic and linguistic correlation of the Kra–Dai-speaking groups in Thailand
Journal of Human Genetics (2015)
