
Abstract
The majority of Xq22 duplications seen in patients with Pelizaeus–Merzbacher disease (PMD) include proteolipid protein 1 (PLP1), the gene responsible for PMD, and neighboring genes. Some cases result from larger duplications up to 7 Mb in size. In comparison, the deletions including PLP1 seen in PMD patients are small. In this study, we present the genetic and clinical information for five female patients with deletions involving the Xq22 region, and review the correlation between the genotype and phenotype. Three of the five patients show similar large deletions (>3 Mb) ranging from Xq22.1 to Xq22.3 and all manifest severe intellectual disability, hypotonia and behavioral abnormalities. The most striking similarity among them are the behavioral problems, including poor eye contact and sleep disturbance. We propose that this represents an emerging distinctive microdeletion syndrome encompassing PLP1 in female patients. The possible candidate region responsible for such distinctive features has been narrowed down to the neighboring region for PLP1, including the interleukin 1 receptor accessory protein-like 2 (IL1RAPL2) gene and the clustered brain expressed X-linked (BEX) genes. The gene(s) responsible for severe neurological features in the patients in this study would be located in the regions proximate to PLP1; thus, males with the deletions involving the gene(s) would be lethal, and finally, the sizes of the deletions in PMD patients would be smaller than those of the duplications.
Similar content being viewed by others

Introduction
Generally, X-chromosome abnormalities result in a more severe phenotype in male patients, whereas females with the same abnormality are often asymptomatic because of heterozygosity of the X chromosome. Mutations and duplications of the proteolipid protein 1 (PLP1) gene located at Xq22 are the major cause of Pelizaeus–Merzbacher disease (PMD; MIM #312080), an X-linked disease that causes congenital leukodystropy associated with spastic paraplegia.1, 2, 3 The mutations and duplications of PLP1 are often inherited from patients’ healthy mothers. Comparatively, loss-of-function mutations, including nonsense mutations and deletions of PLP1, are rare and patients with these types of mutations demonstrate a milder phenotype.4, 5 Female carriers of such mutations can show some mild manifestations, such as mild spasticity,6 without having skewed X-chromosome inactivation (XCI).7 Although a majority of the duplications observed in PMD patients include genes neighboring PLP1 and some of the patients have large duplications, up to 7 Mb in size,8 there are no phenotypic differences between them. In comparison, deletions including PLP1 observed in patients with PMD are smaller, indicating that larger deletions of this region are lethal in males.4
We studied five unrelated females with de novo deletions of Xq22 and three of them presented with severe intellectual disability and behavioral abnormalities, but without any features of PMD. The genotype–phenotype correlation in the five female patients with Xq22 deletions suggests a newly recognized microdeletion syndrome seen in females only, distinct from PMD. The severe presentation of the Xq22 deletions in these females could explain the lack of larger deletions of this region observed in male patients with PMD.
Materials and methods
Patients’ blood samples were obtained with written informed consent based upon approval of each institution’s ethical committee. DNA was extracted from the blood samples and used for subsequent testing. Metaphase spreads were also prepared from blood samples and used for fluorescence in situ hybridization analyses.
Copy number variants were identified using various microarray platforms from Agilent Technologies (Santa Clara, CA, USA), Affymetrix (Santa Clara, CA, USA), and BlueGnome (Cambridge, UK), according to the manufacturer’s protocol (Table 1). Fluorescence in situ hybridization analysis was performed to confirm the results for patient 1, using the following human bacterial artificial chromosomes: RP11-832L2 (Xq22.2: 102 902 650–103 085 315) as a target and RP11-75D20 (Xp22: 13:18 314 474–18 506 931) as a marker; these were selected using the University of California Santa Cruz (UCSC) genome browser (http://www.genome.ucsc.edu). All of the genomic coordinates referred to in this paper are reported in build 19 nomenclature. The data obtained were uploaded onto the web-based database, DECIPHER (http://decipher.sanger.ac.uk/) in accordance with the policy to allow collaboration between centers and help establish new syndromes.
The XCI patterns were analyzed in the patients included in the present study by genotyping the CAG repeats in the androgen receptor gene, using the method described in a previous study.9, 10 Finally, the XCI patterns were classified as random (a ratio higher than 50:50 and lower than 75:25) or skewed (higher than 75:25). Linkage analysis was also performed using the ABI PRIMS Linkage Mapping Set Panel 28 (Life Technologies, Carlsbad, CA, USA) that includes the primer sets of 18 microsatellite markers, including DXS1106 (chrX: 102 731 932–102 732 317) that is located in the deletion region of patient 1; the analysis was performed as described previously.11
Results
Genomic copy number aberrations
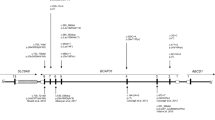
We identified copy number aberrations in the Xq22 region. The five patients with overlapping deletions were identified from five different institutions using the DECIPHER database. The genotype–phenotype correlation was then studied in detail. The details of the Xq22 deletions are summarized in Table 1 and depicted in the genome map (Figure 1). Although patient 5 had an additional duplication on chromosome 11 (chr11: 4 393 604–5 032 655), this was considered to be a possible benign copy number variant as it was inherited from a normal parent.

Results of microarray chromosomal testing and the genome map around Xq22. Red and gray hexagons indicate the deletion regions identified in the patients in this study as well as in previously reported patients. Rectangles indicate the locations of the genes. Genes mentioned in the text are highlighted by red. Possible candidate regions are shown by translucent blue bands.
The absence of an Xq22 deletion was confirmed in the parents of all patients by use of either method (Table 1). Although the linkage study using DXS1106 was uninformative in patient 1, the deletions were confirmed as being de novo using fluorescence in situ hybridization (Supplementary Figure S1). The results of the linkage study showed no contradiction in biological relationship in the family of patient 1 (Supplementary Figure S2). From these results, all of the Xq22 deletions identified in this study were considered to be of de novo origin.
The XCI patterns
The XCI status was analyzed in patients 1, 2, 3 and 5 (Table 1). This analysis was uninformative in patient 1, who was homozygous for the CAG repeat length. Patients 2, 3 and 5 showed skewed XCI patterns.
Clinical reports
The clinical features of the five patients are summarized in Table 2.
Patient 1
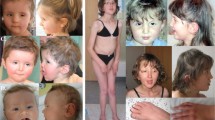
A 3-year-old girl (DECIPHER ID 264512) was born following a pregnancy complicated by polyhydramnios. Transient tachypnea was noted after birth, and the patient was described as being continually irritable when awake and vomiting frequently. Her parents noted poor eye contact and poor social smile at 2–3 months. At 9 months of age, she was diagnosed with developmental delay and underwent a laparoscopic Nissen fundoplication, having previously required nasogastric tube feeding. She had a single seizure in the neonatal period, and although an electroencephalogram demonstrated abnormalities, she had no further episodes. Brain magnetic resonance imaging (MRI) showed no definite abnormality. At her last review, she was underweight (11 kg; <3rd centile) and was still described as irritable and showed severe developmental delay and sleep disturbance. In addition, her eye contact was still poor and she demonstrated self-injury behaviors. There were no involuntary movements, pyramidal signs or stereotypical movements. She has a triangular face with wide nasal bridge, widely spaced eyes, strabismus, a long philtrum and a prominent jaw (Figure 2a).

Facial findings of the patients in this study. (a) Patient 1 at 3 years of age, (b) patient 2 at 7 years of age, (c) patient 3 at 19 years of age and (d) patient 5 at 8 years of age. Similar facial findings including triangle face and a prominent jaw are shared among patients 1–3. Written informed consents for publication of the patients’ photographs were obtained from the guardians of all patients.
Patient 2
A 6-year-old girl (DECIPHER ID 257182) was born at 42 weeks of gestation, after a pregnancy complicated by reduced fetal movements and polyhydramnios. Her birth weight was 4860 g (>97th centile), birth length was 54 cm (>97th centile) and occipitofrontal circumference was 38 cm (>97th centile). Her 37-year-old father is healthy, but her 36-year-old mother suffers from an undiagnosed neuromuscular disease with dominant inheritance and a clear anticipation pattern. The patient has severe developmental delay. She did not manage to support her head until she was over 1-year old. She has a distinct sleeping abnormality, exhibiting hypersomnia, and is described as sleeping ‘most of the time’. There was a history of seizures. An electroencephalogram did not demonstrate any epileptiform activity, but did show increased slow wave activity. An MRI of her brain revealed generalized white matter hypoplasia, most pronounced in the occipital region, delayed myelination, hypoplasia of the corpus callosum and ventriculomegaly. A visual evoked potential indicated neurosensory visual impairment (severe attenuated N1-potential and normal P1-potential).
At present, she still has severe delay and no language skills, and cannot sit or walk. This patient’s appearance is similar to that of patient 1, with a triangular face associated with strabismus and a prominent jaw (Figure 2b). Severe pes equinovarus required surgical correction. She has extremely short feet, corresponding to those of a 3-year-old child.
Patient 3
A 16-year-old girl was born at 43 weeks of gestation by cesarean section. The pregnancy had been complicated by polyhydramnios. Her birth weight was 4820 g (>97th centile). She is the only child of a non-consanguineous couple and there was no significant family history. She developed a social smile at 6 weeks, sat at 7 months and walked independently at 7 years. Her development is severely delayed; she reached the development level of a 7-month-old child when she was 16 months old, and reached a 10-month level when she was three and a half years old. Abnormal self-injury behaviors were noted later, including hitting herself and biting her fingers. She was hypotonic with a mild scoliosis, moderate bilateral hearing loss, constipation and an advanced bone age. Brain MRI examination at 1 year of age showed generalized cerebral atrophy that was slightly worse on the right side. A further MRI at the age of 6 years did not show any specific features, and there was no hypomyelination. Conventional G-banding showed a normal female karyotype.
Currently, she is aphasic and is incontinent. She has fine and slow-growing hair, bi-frontal narrowing, deep-set eyes, a prominent nasal bridge, full upper lip, a prominent jaw, deep palmer creases and prominent volar pads (Figure 2c). Her weight is 56.4 kg (between the 25th and 50th centiles), her height is 146.1 cm (<0.4th centile) and her occipitofrontal circumference is 56 cm (50th–75th centile), indicating short stature.
Patient 4
A 12-month-old girl (DECIPHER ID 265394) was born at term following a normal pregnancy. She is the third child of healthy unrelated parents. During the neonatal period, she had feeding difficulties and her early milestones were delayed. She showed mild motor developmental delay with sitting started from 12 months of age. Because of the bilateral moderate sensorineuronal deafness that was identified at 7 months of age, there was no meaningful word at that time. Social interaction was within normal limit. There was no facial dysmorphism. Behavioral abnormality was not noted.
Patient 5
A 7-year-old girl (DECIPHER ID 253614) weighed 3620 g at term (90–97th centile). From early infancy, she showed psychomotor developmental delay and was only able to sit unsupported at 18 months and walk independently at 6 years. An MRI of the brain showed delayed myelination. She is still aphasic and incontinent. She repeatedly brings her hands to her mouth, hits her own face and appears to have impaired pain perception. She also sleeps poorly. There is no definite facial dysmorphism, apart from strabismus (Figure 2d).
Discussion
In the present study, we conbined the genetic and clinical information of five female patients with deletions within Xq22, and studied the correlation between their genotypes and phenotypes (Figure 1). Three of the five patients (patients 1–3) have similar large deletions, ranging from Xq22.1 to Xq22.3, over 3 Mb in size, and commonly manifested severe intellectual disability, hypotonia and behavioral problems (Table 1). Although patient 2 had a family history of an unknown neuromuscular disease, she did not show any muscular phenotypes and rather showed phenotypes shared with the other two patients (patients 1 and 3). Similar facial features, including a triangular face and a prominent jaw, were also noted among three of the patients (Figures 2a–c). The most striking similarity among them are the behavioral problems, including poor eye contact, self-injuries and sleep abnormality. The shortest region of overlap (SRO) of the deletions in these three patients is between the proximal and the distal breakpoints of patients 2 and 3 respectively (chrX: 101 365 862–103 982 269), and includes PLP1.
Previously, a male patient with Sotos syndrome and Marfanoid features has been reported.12 This patient had a typical 5q35 deletion, which is responsible for the Sotos syndrome, and an additional deletion of Xq22.3. This Xq22.3 deletion was thought to be responsible for his Marfanoid feature. His healthy mother was an asymptomatic carrier of the Xq22.3 deletion. This indicates that the 862 kb deletion at position chrX: 105 167 104–106 028 458 is not pathogenic in females and can be excluded from the region thought to be responsible for the severe neurological manifestations in the patients discussed here. Shimojima et al.12 suggested that their patient’s severe developmental delay may result from a positional effect of the deletion on interleukin 1 receptor accessory protein-like 2 (IL1RAPL2). IL1RAPL2 is a member of the interleukin 1 receptor family, and the similar protein IL1RAPL1, located on Xp21.3–p21.2, is implicated in the development of autism.13 Because the first two exons and the promoter region of this gene are involved in SRO, this gene is one of the candidate genes that may be involved in the symptoms observed in the patients discussed in the present study.
Previously, Grillo et al.14 identified a 1.1-Mb deletion in a female child and her mother with mild intellectual disability that overlaps a portion of the proposed SRO (Figure 1). They discussed the genes responsible for intellectual disability and suggested that some of the nuclear RNA export factor genes that cluster in this region might be responsible for the phenotype. The nuclear RNA export factor 5 (NXF5) gene is the most powerful candidate, as it was disrupted by an inversion in a patient with intellectual disability.15, 16 However, NXF5 is not included in the proposed SRO in this study. Furthermore, this mother and daughter did not show severe intellectual disability and any behavioral abnormalities that are typical of the present three patients (patients 1–3). Therefore, the overlapping region designated as a possible candidate region ‘A’ in Figure 1 can be eliminated.
The deletions in two patients (patients 4 and 5) in the present study are included in the proposed SRO. A 0.25-Mb deletion was identified in patient 4 who showed mild intellectual disability and bilateral sensorineural hearing loss. These are recognized features of Martin–Probst syndrome caused by mutations and deletions in the RAB40A member RAS oncogene family-like (RAB40AL) gene.17 This gene is contained within the deletion in patient 4. This patient did not exhibit hypotonia or behavioral abnormalities. Thus, the deletion region identified in this patient can be excluded from the region thought to be responsible for the severe intellectual disability, hypotonia and behavioral abnormalities observed in the three patients with larger Xq22 deletions (patients 1–3). Furthermore, patient 5 showed a small deletion, 85 kb in size, involving PLP1 and only two further genes, the transmembrane protein 31 (TMEM31) gene and the glycine receptor α4 (GLRA4) gene, about which there is very limited information (Supplementary Table S1). This patient showed severe intellectual disability and behavioral abnormalities, resembling the typical phenotype of the three other patients (patients 1–3).
Although a large number of duplications involving PLP1 have been identified previously, the reciprocal deletion of this region is rarely observed. The previously reported deletions identified in patients with PMD were much smaller than the duplications, with involvement of only two neighboring genes, as seen in patient 5 in the present study (Figure 1 and Supplementary Table S1).4, 18 Presumably, larger deletions of this region would cause lethality or other diverse syndromes.18 Inoue et al.4 observed symptomatic carriers with mild late-onset spastic diplegia. This paradoxical presentation could be explained by mosaicism or random XCI. Figure 1 displays the microdeletions involving PLP1 in the patients in the present study and those reported by Inoue et al.4 and Torisu et al.5 The deletions reported by Inoue et al.4 and Torisu et al.5 were maternally inherited, indicating that females with deletions of this region would not necessarily show severe neurological features. Because the deletion identified in patient 5 overlapps completely with the region reported by Inoue et al.,4 this deletion alone is insufficient to explain her severe intellectual disability and behavior abnormalities. Thus, there must be another reason why patient 5 shows a similar phenotype to the other three patients. We can speculate that there may be very small and very complex chromosomal rearrangements in the neighboring regions of the deletion in patient 5. Microarray technology cannot detect copy-neutral chromosomal rearrangements. Because the clinical features of patient 5 are similar to the other three patients, the critical region for the neurological features common to the other three patients may be disrupted by balanced inversions or translocations in Xq22 region. A coincidental genetic alteration in another chromosomal region is also a possibility. Whole genome sequencing using next-generation sequencing may be the only way to reveal the underlying genomic mechanism in this patient. This is an area for future study. Because the facial features of patient 5 differ from those of the other three patients, the genes responsible for this phenotype can be assumed to be located in SRO, with exception of the deletion region of patient 5.
The possible candidate region for the severe intellectual disability, hypotonia and behavioral abnormalities can therefore be now narrowed down to regions ‘B’ and ‘C’. Region ‘C’ (chrX: 102 993 719–103 982 269) is demarcated by the distal ends of the deletions in patient 3 and Inoue et al. (A).4 Because the only protein-coding gene in this segment is IL1RAPL2, this gene remains a candidate gene. The remaining possible candidate region is ‘B’ that is 724 kb in size (chrX: 102 233 526–102 957 289) and rests between the distal and proximal ends of the deletions in patient 4 and Inoue et al.4 (B), respectively (Figure 1). The brain expressed X-linked (BEX) genes are clustered in this region. This includes BEX1, BEX4, BEX2 and BEX3 (also known as the nerve growth factor receptor-associated protein 1 (NGFRAP1) gene). The Bex gene family members are highly homologous and highly expressed in the brain, but differ in their expression pattern.19 Among them, the most highly validated human Bex ortholog is BEX3 (NGFRAP1) that encodes p75NTR-associated cell death executor (NADE). This interacts with p75 neurotrophin receptor (p75NTR). Neurotrophins are growth factors that play critical roles in the development, maintenance, survival and death of the nervous system.20 Its interactions with p75NTR initiate signal transduction systems that mediate diverse biological functions,21 limiting quantities of neurotrophins during development and controling the numbers of surviving neurons to ensure a match between neurons and the requirement for a suitable density of target innervation.22 Thus, there is a possibility that brain dysfunction may be caused by allelic loss of BEX3 (NGFRAP1) in females. The other BEX genes may also be involved in the neurological features.
As mentioned above, chromosomal deletions encompassing PLP1 are rarely observed in PMD patients, indicating that large deletions in this region in males are likely to be lethal. The sizes of the previously reported deletions are much smaller than the duplications in PMD patients and their carrier mothers and are restricted to the region between regions ‘B’ and ‘C’. This indicates that the gene(s) located on the most proximal neighboring regions of PLP1 would cause lethality in males and cause severe intellectual disability in females, when those were deleted; and finally narrowed the deletion regions in PMD patients. From this perspective, BEX3 (NGFRAP1), located in the region nearest PLP1, is the most powerful candidate gene.
In the present study, three patients showed skewed XCI. Because no patient showed a PMD phenotype, the X chromosome with the deletion involving PLP1 is assumed to be predominantly inactivated in these patients. In spite of this, these patients showed severe intellectual disability. One possible explanation for this phenomenon is that the candidate gene in this region causing severe intellectual disability may escape XCI and that haploinsufficiency of this gene may cause intellectual disability in females. Delayed myelination seen in patient 2 may be because of mildly skewed XCI pattern in this patient. Further cases are likely to be reported in the future and may help clarify the contributions of the genes discussed to the phenotype in these patients. Identifications of nucleotide alterations of these genes would provide further support for this hypothesis.
References
Yamamoto, T. & Shimojima, K. Pelizaeus-Merzbacher disease as a chromosomal disorder. Congenit. Anom. (Kyoto) 53, 3–8 (2013).
Inoue, K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 6, 1–16 (2005).
Shimojima, K., Inoue, T., Hoshino, A., Kakiuchi, S., Watanabe, Y., Sasaki, M. et al. Comprehensive genetic analyses of PLP1 in patients with Pelizaeus-Merzbacher disease applied by array-CGH and fiber-FISH analyses identified new mutations and variable sizes of duplications. Brain Dev. 32, 171–179 (2010).
Inoue, K., Osaka, H., Thurston, V. C., Clarke, J. T., Yoneyama, A., Rosenbarker, L. et al. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am. J. Hum. Genet. 71, 838–853 (2002).
Torisu, H., Iwaki, A., Takeshita, K., Hiwatashi, A., Sanefuji, M., Fukumaki, Y. et al. Clinical and genetic characterization of a 2-year-old boy with complete PLP1 deletion. Brain Dev. 34, 852–856 (2012).
Raskind, W. H., Williams, C. A., Hudson, L. D. & Bird, T. D. Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus-Merzbacher disease. Am. J. Hum. Genet. 49, 1355–1360 (1991).
Soltanzadeh, P., Friez, M. J., Dunn, D., von Niederhausern, A., Gurvich, O. L., Swoboda, K. J. et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul. Disord. 20, 499–504 (2010).
Lee, J. A., Carvalho, C. M. & Lupski, J. R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 (2007).
Hickey, T., Chandy, A. & Norman, R. J. The androgen receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 87, 161–165 (2002).
Shimada, S., Okamoto, N., Ito, M., Arai, Y., Momosaki, K., Togawa, M. et al. MECP2 duplication syndrome in both genders. Brain Dev. 35, 411–419 (2013).
Shimojima, K., Komoike, Y., Tohyama, J., Takahashi, S., Paez, M. T., Nakagawa, E. et al. TULIP1 (RALGAPA1) haploinsufficiency with brain development delay. Genomics 94, 414–422 (2009).
Shimojima, K., Okanishi, T. & Yamamoto, T. Marfanoid hypermobility caused by an 862 kb deletion of Xq22.3 in a patient with Sotos syndrome. Am. J. Med. Genet. A 155A, 2293–2297 (2011).
Piton, A., Michaud, J. L., Peng, H., Aradhya, S., Gauthier, J., Mottron, L. et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 17, 3965–3974 (2008).
Grillo, L., Reitano, S., Belfiore, G., Spalletta, A., Amata, S., Bottitta, M. et al. Familial 1.1 Mb deletion in chromosome Xq22.1 associated with mental retardation and behavioural disorders in female patients. Eur. J. Med. Genet. 53, 113–116 (2010).
Jun, L., Frints, S., Duhamel, H., Herold, A., Abad-Rodrigues, J., Dotti, C. et al. NXF5, a novel member of the nuclear RNA export factor family, is lost in a male patient with a syndromic form of mental retardation. Curr. Biol. 11, 1381–1391 (2001).
Frints, S. G., Jun, L., Fryns, J. P., Devriendt, K., Teulingkx, R., Van den Berghe, L. et al. Inv(X)(p21.1;q22.1) in a man with mental retardation, short stature, general muscle wasting, and facial dysmorphism: clinical study and mutation analysis of the NXF5 gene. Am. J. Med. Genet. A 119A, 367–374 (2003).
Bedoyan, J. K., Schaibley, V. M., Peng, W., Bai, Y., Mondal, K., Shetty, A. C. et al. Disruption of RAB40AL function leads to Martin—Probst syndrome, a rare X-linked multisystem neurodevelopmental human disorder. J. Med. Genet. 49, 332–340 (2012).
Garbern, J. Y. Pelizaeus-Merzbacher disease: genetic and cellular pathogenesis. Cell. Mol. Life Sci. 64, 50–65 (2007).
Alvarez, E., Zhou, W., Witta, S. E. & Freed, C. R. Characterization of the Bex gene family in humans, mice, and rats. Gene 357, 18–28 (2005).
Huang, E. J. & Reichardt, L. F. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736 (2001).
Roux, P. P. & Barker, P. A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 67, 203–233 (2002).
Skaper, S. D. The neurotrophin family of neurotrophic factors: an overview. Methods Mol. Biol. 846, 1–12 (2012).
Acknowledgements
We express our gratitude to the patients and their families for their cooperation. This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas ‘Foundation of Synapse and Neurocircuit Pathology’ from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to TY); a Grant-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare, Japan (to TY); and a Grant-in-Aid for Young Scientists (B) from Japan Society for the Promotion of Science (JSPS) (to KS). MSt thanks ‘Ann Mari & Per Ahlqvist’.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Yamamoto, T., Wilsdon, A., Joss, S. et al. An emerging phenotype of Xq22 microdeletions in females with severe intellectual disability, hypotonia and behavioral abnormalities. J Hum Genet 59, 300–306 (2014). https://doi.org/10.1038/jhg.2014.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.21
Keywords
This article is cited by
-
Confirmation and expansion of the phenotype of the TCEAL1-related neurodevelopmental disorder
European Journal of Human Genetics (2024)
-
Familial 5.29 Mb deletion in chromosome Xq22.1–q22.3 with a normal phenotype: a rare pedigree and literature review
BMC Medical Genomics (2023)
-
Narrowing down the region responsible for 1q23.3q24.1 microdeletion by identifying the smallest deletion
Human Genome Variation (2019)
-
Abnormal mGluR-mediated synaptic plasticity and autism-like behaviours in Gprasp2 mutant mice
Nature Communications (2019)
-
A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies
BMC Neurology (2016)

{kind=link}
{kind=link}