
Abstract
The way in which energy is used in cells is determined under the influence of environmental factors such as nutritional availability. Metabolic adaptation is mainly achieved through the modulation of metabolic gene expression, and may also involve epigenetic mechanisms that enable long-term regulation. Recent studies have identified that nutrients and their metabolites exert an important influence on the epigenome, as they serve as substrates and/or coenzymes for epigenetic-modifying enzymes. Some epigenetic factors have been shown to regulate metabolic genes leading to a shift in energy flow. These findings suggest the concept of metabolism–epigenome crosstalk that may contribute to the formation of a long-term metabolic phenotype. This is particularly relevant to the pathogenesis of obesity and associated metabolic disorders, in which pre- and post-natal nutritional conditions affect disease risks in adulthood. Moreover, most cancer cells exploit metabolic pathways for their hyperproliferative activity, while metabolic misregulation leads to aberrant epigenetic regulation in some cancers. This review explores the possible mechanisms of metabolism–epigenome crosstalk that may facilitate our understanding of physiology and diseases.
Similar content being viewed by others
Introduction
To survive nutritionally threatening conditions, organisms have evolved strategies to exploit limited energy sources for their biological activities. Such strategies are determined at a cellular level, as each cell type harbors differences in energy demands and access to energy sources and oxygen.1 Thus, cells can sense their own energetic status and environment, enabling them to remodel their methods of energy utilization. It is now well accepted that differences in metabolic gene expression largely account for adaptive metabolic changes.2 Recent progress in epigenetics research has suggested the concept that nutrients and their metabolites influence the activities of epigenetic factors, as they often function as coenzymes or substrates for chromatin modifications.3 A plausible biological meaning of this is that nutritional availability affects the epigenomic landscape, which assists the cells in adapting to their environment. Such metabolism–epigenome crosstalk may support the formation of a long-term energy strategy.
The failure to maintain appropriate energy strategies leads to the development of diseases, such as obesity-associated metabolic disorders and cancer.4, 5 Nutritional availability in prenatal and early life affects the metabolic disease risks, indicating that long-term epigenetic alterations may be involved in the development of metabolic diseases.6 Indeed, the impact of caloric excess, deficiencies and imbalances on the epigenome of metabolic organs including skeletal muscle, liver and adipose tissue has been demonstrated in many animal models and some human subjects.7 Although the underlying mechanisms are yet to be revealed, metabolism–epigenome crosstalk is likely to have a central role in connecting dietary habit to disease susceptibility. Cancer cells exhibit extraordinary metabolic flow as they vigorously consume glucose to support rapid cell growth.5 Such metabolism is referred to as the Warburg effect or aerobic glycolysis, meaning a glycolytic-biased metabolism not based on oxygen availability. As epigenetic dysregulation is one of the prominent hallmarks of cancer,8 it is plausible that cancer metabolism could be regulated through epigenetic mechanisms. Conversely, altered metabolic flow has been shown to drive epigenetic misregulation in cancer cells.9, 10
In this review, we will discuss recent progress in understanding how diet and cellular metabolism affects the epigenome, and how epigenetic factors regulate energy metabolism. By focusing on observations in mammalian systems, we will consider the nutritional aspects of lifestyle-associated diseases.
Nutrient sensing by epigenetic factors
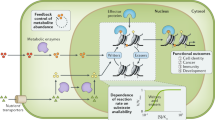
Genomic DNA in eukaryotes is incorporated into a structure called chromatin that enables the efficient packaging of a large DNA in a small nucleus. Minimum structural unit of chromatin is a nucleosome, in which the DNA is wound around a structural core that is constituted of four types of histone proteins, H2A, H2B, H3 and H4. The basic components of epigenetic gene regulation are DNA methylation and histone post-translational modifications, including methylation and acetylation. Combinations of these epigenetic marks primarily reflect the active (accessible) and repressive (inaccessible) chromatin structure according to the ‘histone code hypothesis’.11 In particular, modifications at lysine residues of histone H3 and H4 are often linked to the gene regulation.12 An active chromatin structure is typically decorated with histone acetylation and histone H3 lysine 4 (H3K4) methylation, while repressive marks include hypo-acetylated histones, methylated histone H3 lysine 9 and lysine 27 (H3K9 and H3K27, respectively), H4 lysine 20 (H4K20) and methylated cytosine in DNA (Figure 1). Because of their enzymatic nature, epigenetic factors responsible for the writing and erasing of these marks require substrates and coenzymes that are produced in cells from nutrients; therefore, important epigenetic marks should theoretically be influenced by the cellular metabolic state (Figure 1).

Metabolism–epigenome crosstalk. Environmental factors such as nutrition and oxygen availability affect metabolic pathways resulting in altered cellular concentrations of metabolites. These metabolites include acetyl-CoA, NAD+, S-adenosyl-methionine (SAM), α-KG and flavin adenosine dinucleotide (FAD), which serve as substrates or coenzymes for epigenetic-modifying enzymes. Activities of these enzymes contribute to the chromatin structure, which in turn modulates the expression of metabolic genes. Combinations of histone methylation/acetylation and DNA cytosine methylation represent the active and repressive chromatin structures. A full color version of this figure is available at the Journal of Human Genetics journal online.
DNA or histone methylations require S-adenosyl-methionine as a methyl group donor, which is synthesized from diet-derived methionine through the one-carbon cycle. This process also involves folate, choline, betaine and vitamin B12, and the dietary intake of these nutrients affects S-adenosyl-methionine production.13 Early studies on the agouti viable yellow (Avy) allele in mice established the concept of how diet affects S-adenosyl-methionine production and subsequent DNA methylation events (reviewed by Bernal and Jirtle14). The Avy allele contains an insertion of a retrotransposon, intracisternal A-particle, in the upstream of the agouti locus encoding agouti signaling protein. The expression of this gene is highly dependent on the DNA methylation status of intracisternal A-particle: when highly methylated, the use of wild-type promoter enables a low level and a hair cycle-specific expression, while lower methylation level activates a cryptic promoter, which results in a high and disordered expression. The expression of this locus is directly associated with the coat color of mice: wild-type expression results in dark color (pseudoagouti), while the expression from cryptic promoter leads to yellow. Thus, the coat color, at least in part, reflects the epigenotype in these mice. In normal conditions, Avy mice exhibit varying levels of DNA methylation at Avy locus, with the coincident variation of the coat color. However, when pregnant mice are fed with a methyl donor-supplemented diet, the frequency of offsprings with darker coat color increases significantly.15 These studies on Avy mice provided important insights that the nutrition, the epigenotype and the phenotype are interconnected.
To date, 12 histone demethylases have been identified in mammals, all of which are categorized into either amine oxidase or jumonji-containing dioxygenase families.16 Lysine-specific demethylases (LSD) 1 and 2 comprise the amine oxidase family, which require flavin adenosine dinucleotide as a coenzyme.17, 18 Flavin adenosine dinucleotide (FAD) is mainly produced in the mitochondria from dietary riboflavin,19 and is converted into its reduced form, FADH2, through metabolic processes such as fatty acid oxidation and succinate dehydrogenation in the tricarboxylic acid cycle, suggesting that cellular metabolic flow affects the activities of LSD1 and 2. Jumonji-containing dioxygenases are also potentially sensitive to metabolic conditions as they require α-ketoglutarate (α-KG), a tricarboxylic acid cycle intermediate, and oxygen.20 Furthermore, hydroxylation of methylated DNA is thought to be a transition step for DNA demethylation, and the responsible enzymes, ten-eleven translocation (TET)-1, -2 and -3, are also α-KG-dependent dioxygenases.21
Identification of the sirtuin family of proteins as nicotinamide adenine dinucleotide (NAD)+-dependent histone deacetylases (HDACs) led to the notion that histone acetylation status could be affected by cellular metabolism.22 Acetyl coenzyme A (Acetyl-CoA), produced from glucose or fatty acids, is required as an acetyl group donor for histone acetylation. Wellen et al.23 found that ATP citrate lyase, an enzyme that converts citrate into acetyl-CoA, was abundantly present in the nucleus. Glucose deprivation reduced the size of the acetyl-CoA pool in an ATP citrate lyase-dependent manner, with decreased histone acetylation and transcription of a subset of genes.
Epigenetic modifications associated with metabolic disorders
Obesity and consequent insulin resistance represent typical examples of a cellular failed energy strategy. It is well documented that nutrient- and hormone-driven transcription regulators greatly contribute to the disrupted metabolic gene expression under caloric excess.2 In particular, the downregulation of mitochondrial respiration genes in obese and diabetic subjects is linked to the mitochondrial dysfunction that leads to compromised systemic energy homeostasis.24, 25, 26 Since the early prediction by Hales and Barker,27 undernutrition during the prenatal and childhood periods have been associated with the risks of type II diabetes mellitus and cardiovascular diseases.27, 28 This evidence suggests the existence of an underlying epigenetic mechanism that explains the long-term influence of nutritional intake on mitochondrial dysfunction in metabolic syndrome.
The reduction of fatty acid β-oxidation is one of the important features of mitochondrial dysfunction in the skeletal muscle, which is closely associated with the insulin resistance in metabolic diseases.29 A report by Barres et al.30 showed increased DNA methylation on the promoter of the peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α gene in the skeletal muscle of type II diabetes mellitus patients, in which the expression of this gene was downregulated. As PGC-1α is an integrative transcriptional regulator of oxidative metabolism including fatty acid oxidation and mitochondrial biogenesis,31 DNA methylation-dependent repression of this gene might explain the mechanism of mitochondrial dysfunction. The paper also demonstrated that this methylation pattern could be reproduced in vitro by exposing myocytes to free-fatty acids, and that the methylation was dependent on DNMT3B (DNA (cytosine-5-)-methyltransferase 3 beta). Another report analyzed PGC-1α promoter methylation in young healthy men with low- and normal- birth- weights.32 The skeletal muscle of low birth weight individuals harbored higher methylation frequencies than normal birth weight individuals, and was more susceptible to insulin resistance after short-term overfeeding. This implies that the perinatal metabolic condition is responsible for the epigenetic changes that lead to an increased sensitivity to caloric excess later in life. Liver is another important source of oxidative metabolism, which contributes to the systemic metabolic homeostasis. Calorie overload leads to the accumulation of hepatic lipids, resulting in the development of nonalcoholic fatty liver disease including nonalcoholic steatohepatitis, which is associated with mitochondrial dysfunction.33 The decreased mitochondrial DNA content in nonalcoholic fatty liver disease patients was coincident with increased PGC-1α promoter methylation, suggesting that an epigenetic event might be involved in mitochondrial dysfunction in the liver.34 In addition, PGC-1α is a crucial regulator of gluconeogenesis in the liver, indicating that hepatic glucose production might also be regulated epigenetically. It should be noted that while studies on human subjects highlight the important contribution of DNA methylation, animal experiments have revealed the impact of diet on histone methylation and acetylation, many of which are indicative of metabolic outcomes (reviewed in detail by Jimenez-Chillaron et al.7).
Neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases are also under the influence of dietary lifestyle, and are associated with mitochondrial dysfunction with reduced respiratory gene expression.35, 36, 37 Thus, epigenetic disruption of these genes may represent a common feature of lifestyle-associated diseases.
Possible epigenetic mechanisms of diet-associated metabolic diseases
Although experimental and clinical evidence strongly suggest the occurrence of epigenomic aberration in metabolic syndrome, the underlying mechanisms of how epigenetic factors are recruited to and dissociated from the metabolic genes are poorly understood. Nevertheless, some in vitro and animal studies predict the possible pathways that link specific epigenetic factors to metabolic dysfunctions.
Excess lipid accumulation in the adipose tissues under calorie overload triggers metabolic dysfunction in many organs, leading to the development of type II diabetes mellitus and cardiovascular diseases. Thus, the balance between energy storage and expenditure in the adipocytes must be tightly regulated in order to maintain systemic energy homeostasis. There are two distinct categories of adipose tissues in mammalian body.38 White adipose tissues are primarily responsible for the storage of excess energy in the form of triglycerides, whereas brown adipose tissue consumes a large amount of glucose and fatty acids for thermogenesis under cold exposure. A pioneering work presented a possible epigenetic mechanism of the adipose tissue homeostasis by demonstrating that the histone acetyltransferase, CREB-binding protein, was involved in the differentiation and the fat accumulation of white adipocytes.39 Later studies have identified the regulation of DNA/histone methylation in relation to the energy metabolism in adipocytes and some other cell types. For example, a paper by Tateishi et al.40 demonstrated that mutant mice lacking Jhdm2a, a jumonji-type histone H3K9 demethylase, developed obesity and hyperlipidemia when fed a normal diet. These mice exhibited impaired thermogenesis in response to adrenergic stimuli in brown adipose tissue, which may partially explain the cause of the metabolic imbalance. Mechanistically, the uncoupling protein-1 (Ucp-1) gene, a critical regulator of thermogenesis in brown adipose tissue, was downregulated with increased H3K9 methylation and with reduced occupancy of a transcription factor, peroxisome proliferator-activated receptor-α, on the promoter. JHDM2A has also been shown to regulate glucose- and lipid-handling genes in white adipose tissue in mice.41 Thus, this demethylase may be an integrative regulator of energy balance in adipose cells.
Kamei et al.42 reported the increased expression of Dnmt3a, a DNA methyltransferase, in white adipose tissue in mice with diet-induced obesity. Moreover, the adipose-specific transgenic expression of Dnmt3a in the mice enhanced the expression of inflammation-associated genes such as monocyte chemotactic protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α) in obese adipose tissue, suggesting its relevance to chronic inflammation in obesity. A different report revealed the contribution of de novo DNA methylation to metabolic transition during lactation and weaning periods.43 The rate of hepatic lipogenesis is low when feeding on a mother’s milk enriched with lipids, and it rises after weaning. In the neonatal mouse liver, DNMT3B methylates the promoter of the glycerol-3-phosphate acyltransferase 1 (Gpat1) gene, inhibiting the binding of a key transcription factor, SREBP-1c. After weaning, the Gpat1 promoter becomes unmethylated and is susceptible to SREBP-1c-mediated transcription. It is of great interest to see how differences in dietary composition affect the expression and activity of DNMTs and consequent DNA methylation patterns. A report by Brasacchio et al.44 showed that the glucose-dependent induction of NF-κB/p65 gene expression coincided with Set7-dependent histone H3 lysine 4 methylation, and was counteracted by LSD1. The expression of NF-κB/p65 in vascular endothelial cells was increased by exposure to high levels of glucose, and persisted for up to 6 days.44, 45 These observations may help us understand how metabolic memory can be established and maintained.
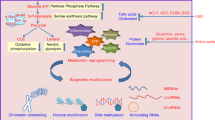
We recently found that LSD1 suppresses energy expenditure in white adipocytes (Figure 2).46 The inhibition of LSD1 function by either RNA interference or selective inhibitor drugs resulted in the activation of mitochondrial respiration, as assessed by the oxygen consumption rate and the inner membrane potential. This metabolic remodeling was accompanied with the increased expression of energy expenditure-associated genes, including PGC-1α, pyruvate dehydrogenase kinase (PDK4) and adipose triglyceride lipase (ATGL), and the enrichment of methylated histone H3 lysine 4 on their promoters. Interestingly, the gene repressive function of LSD1 was emphasized when excess lipid accumulation was induced by insulin stimulation in vitro and by high-fat feeding in vivo. Moreover, LSD1 exerted its gene regulatory function depending on intracellular flavin adenosine dinucleotide production. Thus, LSD1 contributes to the metabolic epigenome, which facilitates energy storage in white adipocytes, depending on the caloric environment and intracellular metabolic condition.

FAD-dependent LSD1 regulates energy expenditure. LSD1 epigenetically represses energy expenditure genes in white adipose cells. A full color version of this figure is available at the Journal of Human Genetics journal online.
Although epigenetic factors potentially affect chromatin structure in a genome-wide manner, these observations suggest that the modulation of a single epigenetic factor can impact cellular metabolic flow and systemic energy homeostasis. They also suggest that the function of epigenetic factors as metabolic regulators depends on the cell type, the environmental condition (for example, nutrition) and co-operating transcription factors.
Epigenetic regulation of cancer metabolism
Cancer cells rely on the glycolytic pathway for their energy production regardless of the availability of oxygen.5 Such metabolic remodeling enables the efficient production of biochemical materials including nucleotides and lipids, supporting their rapid proliferation. As epigenetic dysregulation contributes to cancer development, it is plausible that epigenetic changes of energy metabolism genes trigger glycolytic bias during tumorigenesis.47 Indeed, the mRNA expression of fructose-1,6-bisphosphatase, an inhibitor of glycolysis, is downregulated by promoter hypermethylation in hepatic and colon cancers.48 Although the epigenomic landscape of metabolic gene regulation in cancer has not been fully characterized, some studies provide mechanistic insights into how metabolism and epigenetic factors interact to co-ordinate the Warburg effect.
A recent study identified SIRT6, a sirtuin family HDAC, as a tumor suppressor that regulates aerobic glycolysis.49 Sirt6-KO mouse embryonic fibroblasts showed glycolysis-biased metabolism in vitro, and were highly tumorigenic compared with wild-type cells. SIRT6 repressed glycolytic and ribosomal protein genes by reducing H3K56 acetylation levels in tumor cells. Importantly, the expression of SIRT6 was downregulated in pancreatic cancer and colon adenocarcinoma, indicating that SIRT6 loss drives cancer-type energy metabolism in human cancers.
Colonocytes utilize butyrate, a short-chain fatty acid produced in the colon from dietary fiber through bacterial fermentation, as an energy source, as they can metabolize it in the mitochondria, producing acetyl-CoA that enters the tricarboxylic acid cycle.50 In addition, butyrate is well known as an HDAC inhibitor,51 suggesting that the function of butyrate might represent another example of nutrient-epigenome communications. In terms of colon cancer, although butyrate inhibits cell growth and induces apoptosis of cancerous colonocytes in vitro,52 controversial results have been presented on whether diet-derived butyrate could be cancer preventive in vivo.53 Recently, Donohoe et al.54 demonstrated that physiological doses of butyrate direct aberrant epigenomic and transcriptomic profiles in cancerous colonocytes. Because of the glycolytic shift, the capacity of colon cancer cells to metabolize butyrate is limited, allowing large fractions of butyrate to serve as an HDAC inhibitor in the nucleus. Interestingly, under the Warburg effect, colon cancer cells show high expression of ATP citrate lyase, which contributes to the conversion of butyrate to acetyl-CoA as a substrate for histone acetylation in the nucleus. Thus, colon cancer cells harbor the decreased HDAC activity and the increased level of acetyl group donor, both of which may promote global histone acetylation. Indeed, the epigenetic impact of butyrate was selectively exerted against the genes involved in cell cycle regulation, which showed increased expression and promoter histone acetylation in a butyrate-dependent manner. These findings are particularly important because they raise the possibility that the Warburg effect might be a cause, rather than a consequence, of aberrant gene expression in cancer cells.
A series of studies on isocitrate dehydrogenase (IDH) 1 and 2 gene-mutated cancers illustrates how misdirected metabolic flow influences the global epigenome in cancer. Amino acid substitutions of IDH1 and 2 are frequently found in cancers, especially in the glioma and acute myeloid leukemia.55, 56 Although wild-type IDH produces α-KG from isocitrate as a tricarboxylic acid cycle component, cancer-associated mutants further metabolize α-KG into the (R)-enantiomer of 2-hydroxyglutarate ((R)-2-HG), which normally exists at very low concentrations in cells.57 As α-KG is an essential coenzyme for jumonji-histone demethylases and the TET family of DNA hydroxylases and as (R)-2-HG has been shown to inhibit these enzymes,58, 59 recent studies have focused on the ability of IDH mutants to drive epigenetic aberrations. Acute myeloid leukemia with IDH1/2 mutations show DNA methylation profiles resembling those of TET2 mutants.60 The contribution of IDH-dependent metabolism to TET2 function was also identified in melanoma cells, in which both IDH2 and TET2 genes were downregulated with a reduced global hydroxylation of methylated cytosines.61 Overexpression of these genes suppressed tumor growth, indicating their roles as tumor suppressors. Recently, (R)-2-HG was verified as an oncometabolite that drives leukemogenesis.62 Again, TET2 was identified as a key effector, as the knockdown of TET2 abolished the oncogenic effect of (R)-2-HG. Although these findings delineate the disruption of the DNA methylation pattern through the (R)-2-HG/TET-mediated pathway in IDH-mutant cancer, it is likely that jumonji demethylases are also involved, as histone methylation changes are observed in these cancers.9
Conclusion
As described above, recent progress in epigenetics research has identified how metabolites affect the function of epigenetic factors to regulate gene expression. The next challenge in the field is to understand how individual metabolite-epigenetic factor communication co-ordinates to form a particular epigenotype that leads to a biological outcome. A reasonable approach towards this end may be to investigate why selective sets of genes are affected by metabolites, when the whole genome should potentially be susceptible to epigenetic changes.23, 46 Testing the possible involvement of nutrient- and/or hormone-driven signaling pathways and transcription factors may help to elucidate this point. In terms of metabolic diseases, we lack the knowledge on whether the diet affects the availability of the nutrient-derived epigenetic cofactors (for example, methionine, riboflavin and so on) in the cells to drive aberrant epigenetic regulation. The investigation on this point would provide mechanistic insights into how specific epigenetic factors might influence the pathogenesis of metabolic diseases.
The long-term effects of metabolic epigenome formation should also be characterized to understand how dietary habits affect the risks of lifestyle-associated diseases, including metabolic syndrome and cancer. The first approach for this may be to identify the fraction of cells that harbor epigenetic alterations in response to dietary composition in vivo, as such cells must somehow maintain the acquired epigenotype to support the long-term environmental effect. In conclusion, research into metabolism–epigenome crosstalk would focus on the general area of how the environment influences health and disease.
References
Mohyeldin, A., Garzon-Muvdi, T. & Quinones-Hinojosa, A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7, 150–161 (2010).
Desvergne, B., Michalik, L. & Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 86, 465–514 (2006).
Kaochar, S. & Tu, B. P. Gatekeepers of chromatin: small metabolites elicit big changes in gene expression. Trends. Biochem. Sci. 37, 477–483 (2012).
Tseng, Y. H., Cypess, A. M. & Kahn, C. R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug. Discov. 9, 465–482 (2010).
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell 21, 297–308 (2012).
Hanson, M. A. & Gluckman, P. D. Developmental origins of health and disease: new insights. Basic. Clin. Pharmacol. Toxicol. 102, 90–93 (2008).
Jimenez-Chillaron, J. C., Diaz, R., Martinez, D., Pentinat, T., Ramon-Krauel, M., Ribo, S. et al. The role of nutrition on epigenetic modifications and their implications on health. Biochimie 94, 2242–2263 (2012).
Suva, M. L., Riggi, N. & Bernstein, B. E. Epigenetic reprogramming in cancer. Science 339, 1567–1570 (2013).
Turcan, S., Rohle, D., Goenka, A., Walsh, L. A., Fang, F., Yilmaz, E. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H. et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338 (2012).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Latchman, D. S. Gene control, (Garland Science, 2010).
Anderson, O. S., Sant, K. E. & Dolinoy, D. C. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 23, 853–859 (2012).
Bernal, A. J. & Jirtle, R. L. Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res. A Clin. Mol. Teratol. 88, 938–944 (2010).
Wolff, G. L., Kodell, R. L., Moore, S. R. & Cooney, C. A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J. 12, 949–957 (1998).
Pedersen, M. T. & Helin, K. Histone demethylases in development and disease. Trends. Cell. Biol. 20, 662–671 (2010).
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Karytinos, A., Forneris, F., Profumo, A., Ciossani, G., Battaglioli, E., Binda, C. et al. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 284, 17775–17782 (2009).
Barile, M., Brizio, C., Valenti, D., De Virgilio, C. & Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 267, 4888–4900 (2000).
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Wu, H. & Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452 (2011).
Houtkooper, R. H., Pirinen, E. & Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell. Biol. 13, 225–238 (2012).
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R. & Thompson, C. B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Mootha, V. K., Lindgren, C. M., Eriksson, K. F., Subramanian, A., Sihag, S., Lehar, J. et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Patti, M. E., Butte, A. J., Crunkhorn, S., Cusi, K., Berria, R., Kashyap, S. et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl Acad. Sci. USA 100, 8466–8471 (2003).
Kelley, D. E., He, J., Menshikova, E. V. & Ritov, V. B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950 (2002).
Hales, C. N. & Barker, D. J. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35, 595–601 (1992).
Gluckman, P. D., Hanson, M. A., Buklijas, T., Low, F. M. & Beedle, A. S. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat. Rev. Endocrinol. 5, 401–408 (2009).
Turner, N. & Heilbronn, L. K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 19, 324–330 (2008).
Barres, R., Osler, M. E., Yan, J., Rune, A., Fritz, T., Caidahl, K. et al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell. Metab. 10, 189–198 (2009).
Houten, S. M. & Auwerx, J. PGC-1alpha: turbocharging mitochondria. Cell 119, 5–7 (2004).
Brons, C., Jacobsen, S., Nilsson, E., Ronn, T., Jensen, C. B., Storgaard, H. et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 95, 3048–3056 (2010).
Sanyal, A. J., Campbell-Sargent, C., Mirshahi, F., Rizzo, W. B., Contos, M. J., Sterling, R. K. et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192 (2001).
Sookoian, S., Rosselli, M. S., Gemma, C., Burgueno, A. L., Fernandez Gianotti, T., Castano, G. O. et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 52, 1992–2000 (2010).
Mattson, M. P. Gene-diet interactions in brain aging and neurodegenerative disorders. Ann. Intern. Med. 139, 441–444 (2003).
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L. et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2, 52ra73 (2010).
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T. & Brinton, R. D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 106, 14670–14675 (2009).
Gesta, S., Tseng, Y. H. & Kahn, C. R. Developmental origin of fat: tracking obesity to its source. Cell 131, 242–256 (2007).
Yamauchi, T., Oike, Y., Kamon, J., Waki, H., Komeda, K., Tsuchida, A. et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat. Genet. 30, 221–226 (2002).
Tateishi, K., Okada, Y., Kallin, E. M. & Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761 (2009).
Inagaki, T., Tachibana, M., Magoori, K., Kudo, H., Tanaka, T., Okamura, M. et al. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14, 991–1001 (2009).
Kamei, Y., Suganami, T., Ehara, T., Kanai, S., Hayashi, K., Yamamoto, Y. et al. Increased expression of DNA methyltransferase 3a in obese adipose tissue: studies with transgenic mice. Obesity (Silver Spring) 18, 314–321 (2010).
Ehara, T., Kamei, Y., Takahashi, M., Yuan, X., Kanai, S., Tamura, E. et al. Role of DNA methylation in the regulation of lipogenic glycerol-3-phosphate acyltransferase 1 gene expression in the mouse neonatal liver. Diabetes 61, 2442–2450 (2012).
Brasacchio, D., Okabe, J., Tikellis, C., Balcerczyk, A., George, P., Baker, E. K. et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236 (2009).
El-Osta, A., Brasacchio, D., Yao, D., Pocai, A., Jones, P. L., Roeder, R. G. et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 205, 2409–2417 (2008).
Hino, S., Sakamoto, A., Nagaoka, K., Anan, K., Wang, Y., Mimasu, S. et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 3, 758 (2012).
Wang, X. & Jin, H. The epigenetic basis of the Warburg effect. Epigenetics 5, 566–568 (2010).
Chen, M., Zhang, J., Li, N., Qian, Z., Zhu, M., Li, Q. et al. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS One 6, e25564 (2011).
Sebastian, C., Zwaans, B. M., Silberman, D. M., Gymrek, M., Goren, A., Zhong, L. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199 (2012).
Donohoe, D. R., Garge, N., Zhang, X., Sun, W., O’Connell, T. M., Bunger, M. K. et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell. Metab. 13, 517–526 (2011).
Davie, J. R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 133, 2485S–2493S (2003).
Andriamihaja, M., Chaumontet, C., Tome, D. & Blachier, F. Butyrate metabolism in human colon carcinoma cells: implications concerning its growth-inhibitory effect. J. Cell. Physiol. 218, 58–65 (2009).
Lupton, J. R. Microbial degradation products influence colon cancer risk: the butyrate controversy. J. Nutr. 134, 479–482 (2004).
Donohoe, D. R., Collins, L. B., Wali, A., Bigler, R., Sun, W. & Bultman, S. J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell. 48, 612–626 (2012).
Ichimura, K. Molecular pathogenesis of IDH mutations in gliomas. Brain. Tumor. Pathol. 29, 131–139 (2012).
Rakheja, D., Konoplev, S., Medeiros, L. J. & Chen, W. IDH mutations in acute myeloid leukemia. Hum. Pathol. 43, 1541–1551 (2012).
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Chowdhury, R., Yeoh, K. K., Tian, Y. M., Hillringhaus, L., Bagg, E. A., Rose, N. R. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO. Rep. 12, 463–469 (2011).
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell 19, 17–30 (2011).
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell 18, 553–567 (2010).
Lian, C. G., Xu, Y., Ceol, C., Wu, F., Larson, A., Dresser, K. et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146 (2012).
Losman, J. A., Looper, R. E., Koivunen, P., Lee, S., Schneider, R. K., McMahon, C. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013).
Acknowledgements
SH is supported by a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science, and by the Nakatomi Foundation. MN is supported by a Grant-in-Aid for Scientific Research on Innovative Areas (3307), from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by the Japan Science and Technology Agency (CREST), and by a grant from the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hino, S., Nagaoka, K. & Nakao, M. Metabolism–epigenome crosstalk in physiology and diseases. J Hum Genet 58, 410–415 (2013). https://doi.org/10.1038/jhg.2013.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.57
Keywords
This article is cited by
-
Rate of entropy model for irreversible processes in living systems
Scientific Reports (2017)
