
Abstract
Genomic imprinting is an important epigenetic factor in complex traits study, and there has recently been considerable interest in association study for quantitative traits by incorporating imprinting. However, these methods need the assumptions of Hardy–Weinberg equilibrium or only use information from families with one child. In this paper, by taking imprinting into account and making no assumption about the distribution of the quantitative traits, we propose two novel classes of Q-C-TDTI(c) and Q-C-MAX(c) family-based association tests for quantitative traits. The tests flexibly accommodate family data with missing parental genotype and with multiple siblings. Q-C-TDTI(c) is derived from a two-stage analysis, where in the first stage Q-C-PAT(c) is applied to test for imprinting effects and in the second stage we select the most appropriate statistic among three transmission disequilibrium tests for association according to the finding from Q-C-PAT(c). Another proposed Q-C-MAX(c) approach takes the maximum of the three statistics. Compared with the existing alternative methods, the simulation results demonstrate that the two proposed tests are robust to population stratification and have better performance for testing association under various scenarios. Further, the powerful and versatile Q-C-TDTI(c) test is applied to analyze Framingham Heart Study data.
Similar content being viewed by others



Introduction
For a diallelic marker locus, the transmission disequilibrium test (TDT) based on case–parent trios is a simple and powerful tool in fine mapping for dichotomous traits.1 As such, TDT has further been extended to test for the association for quantitative traits in the literature.2, 3, 4, 5, 6, 7, 8, 9, 10 For example, Abecasis et al.3 proposed a genotype decomposition method, the quantitative TDT (QTDT), which allows for an arbitrary number of children in each nuclear family and is robust to population stratification. However, QTDT needs the assumption that the quantitative traits under study are normally distributed, which may be violated in practice. Relaxing the assumption on the distribution of the quantitative traits, Sun et al.4 suggested a class of association tests based on the nonparametric extensions of the TDT-type tests for quantitative traits, including the class of T2(c) tests in families with both parents genotyped (complete nuclear families) and the class of T1(c) tests in families each with only one parent genotyped (incomplete nuclear families), where c is any constant. Kistner and Weinberg8, 9 proposed a quantitative polytomous logistic regression to test for association and linkage between markers and quantitative traits, which is termed QPL in this paper for simplicity.
Genomic imprinting is a genetic phenomenon in which the expression level of a gene depends on whether it is paternally or maternally inherited. To date, 72 imprinted genes in human have been reported (http://igc.otago.ac.nz). There has been considerable interest in the association study for quantitative traits by incorporating imprinting effects. Abecasis et al.3 suggested a global test for both the main allelic effect and the imprinting effect implemented in QTDT package (http://www.sph.umich.edu/csg/abecasis/QTDT/), which is denoted by QTDT-G in this paper. However, this method requires the identical by descent information from families and it only accommodates complete nuclear families with both parents available. Van den Oord5 developed a finite mixture model to test for linkage disequilibrium (LD), maternal effects and parent-of-origin effects for complete and incomplete nuclear families. The method is computationally intensive when there are multiple children in a family and no standard software is available.11 Furthermore, both the above-mentioned methods need the normality assumption on the distribution of the quantitative traits. On the other hand, making no assumption on the distribution of quantitative traits, Kistner et al.12 further extended the original QPL test for maternally mediated effects and parent-of-origin effects. However, it can only accommodate families with one child. Recently, methods based on the so-called measured genotype approach and using mixed models for the analysis of association of quantitative traits, including imprinting effects, have been proposed.13, 14 Such methods assume Hardy–Weinberg equilibrium for haplotypes in the identification of parental origin of alleles, and thus may generate false positives in the presence of population stratification.
In this paper, by taking imprinting effects into account and making no assumption about the distribution of the quantitative traits, we propose two novel classes of association tests, Q-C-TDTI(c) and Q-C-MAX(c). The proposed tests are featured in targeting association mapping of quantitative trait loci, and accommodating family data with different combinations of parent–child trios, parent–child pairs with missing parental genotypes and nuclear families with multiple siblings. The Q-C-TDTI(c) test is derived from a two-stage analysis, where in the first stage we apply Q-C-PAT(c)11 to test for imprinting effects, and in the second stage we select the most appropriate statistic among three TDT-type tests, developed on the extensions of the association tests proposed for qualitative traits in previous studies,15, 16 for association according to the finding from Q-C-PAT(c). The alternative Q-C-MAX(c) approach is also proposed, which takes the maximum of the three statistics. Extensive simulation studies are conducted to evaluate the validity and performance of the proposed tests in the population stratification model. Simulation results show that the proposed statistics control the size well under the null hypothesis of no association and Q-C-TDTI(c) is more powerful than Q-C-MAX(c), QTDT-G and QPL tests under the alternative. As such, only the powerful Q-C-TDTI(c) test is used for the analysis of the Framingham Heart Study data.
Materials and methods
Notations and assumptions
Consider a quantitative trait of interest with mean trait value μ. Assume that the quantitative trait locus (QTL) responsible for the trait has a variant allele D1 and a normal one D2. To incorporate the imprinting information into association analysis, let the mean trait values of the four ordered genotypes D1/D1, D1/D2, D2/D1 and D2/D2 at the QTL be, respectively, designated as

where a is the additive effect of allele D1 and d=(d1+d2)/2 is the dominance effect. Without loss of generality, we assume that the left allele of each genotype is paternal and the right one is maternal. Then, the degree of imprinting effects I can be measured by I=(d1−d2)/2. In the absence of dominance effect (d=0), |I| takes value from the interval [0, a]. In particular, I=±a denotes complete imprinting effect and I=0 represents no imprinting effects. Further, we suppose that the diallelic marker locus under study has alleles M1 and M2. For a parent–child trio, let F, M and C be the number of copies of allele M1 carried by the father, mother and child, respectively. As such, F, M and C take possible values of 0, 1 or 2. Just like Kistner and Weinberg8, 9 and He et al.,11 mating symmetry is assumed in parental generation within each mating type; that is, Pr[F=f, M=m]=Pr[F=m, M=f] for all f, m=0, 1, 2. We further assume that without loss of generality the target allele M1 is in positive LD with the allele D1 at QTL when association exists, and there is no maternal effect of the QTL.
Tests for complete nuclear families
Suppose that we have n2 complete nuclear families with both parents available. Let li denote the number of children and Qij be the trait value of the jth child in the ith family, i=1,…, n2, j=1,…, li. Define sij=Tij−NTij, where Tij and NTij denote the numbers of allele M1 being transmitted and not being transmitted from the heterozygous parents in the ith family to his/her jth child, respectively. For any constant c, the following class of tests proposed by Sun et al.4 was proposed to test for association at the QTL:

where


Here  is the unbiased estimate of the variance of s2(c) conditional on children's trait values and the corresponding parental genotypes under the null hypothesis H0 of no association between alleles M1 and D1.
is the unbiased estimate of the variance of s2(c) conditional on children's trait values and the corresponding parental genotypes under the null hypothesis H0 of no association between alleles M1 and D1.
Note that incorporating the information on imprinting effects into association analysis can improve the test power for association. Following the previous studies for qualitative traits,15, 16 we first stratify the transmission/nontransmission numbers of allele M1 from heterozygous parents to their children into the paternal and maternal cases. Specifically, define Tpij (Tmij) and NTpij (NTmij) as the numbers of allele M1 being transmitted and not being transmitted from the heterozygous father (mother) in the ith family to his (her) jth child, respectively. For qualitative traits, Hu et al.15 developed the tests TDTp and TDTm based on the paternal and maternal stratification of the conventional TDT. The generalized versions of TDTp and TDTm for families with multiple siblings are expressed as  and
and  , respectively, where
, respectively, where  and
and  , and
, and  and
and  are the unbiased estimates of the variances of s2p and s2m under H0, respectively. It has been shown by Hu et al.15 that TDTp and TDTm are more powerful than the traditional TDT for testing for association in the presence of maternal and paternal imprinting effects, respectively. Inspired by the research work, we propose the paternal and maternal versions of the T2(c), which are formulated as
are the unbiased estimates of the variances of s2p and s2m under H0, respectively. It has been shown by Hu et al.15 that TDTp and TDTm are more powerful than the traditional TDT for testing for association in the presence of maternal and paternal imprinting effects, respectively. Inspired by the research work, we propose the paternal and maternal versions of the T2(c), which are formulated as  and
and  , respectively, where
, respectively, where  and
and  .
.  and
and  are the unbiased estimates of the variances of s2p(c) and s2m(c) under H0, respectively. It is proved in Supplementary Appendix I that T2p(c) and T2m(c) have approximate standard normal distributions under H0.
are the unbiased estimates of the variances of s2p(c) and s2m(c) under H0, respectively. It is proved in Supplementary Appendix I that T2p(c) and T2m(c) have approximate standard normal distributions under H0.
T2m(c), T2(c) and T2p(c) are expected to be the optimal association tests in the case of paternal imprinting, no imprinting and maternal imprinting, respectively. Thus, we propose a two-stage analysis, where we identify the underlying imprinting effect in the first stage followed by testing for association using the optimal T2(c)-type test in the second stage. Specifically, in the first stage, the test statistic  (He et al., 2011) is used to classify the imprinting effects. In the formula,
(He et al., 2011) is used to classify the imprinting effects. In the formula,  is an unbiased estimate of the variance of sI(c) under no imprinting effects and sI(c) is given by
is an unbiased estimate of the variance of sI(c) under no imprinting effects and sI(c) is given by  with sIij=, where I{comparison statement} is 1 when the comparison statement holds and is 0 otherwise. The imprinting effect is classified into paternal imprinting effect if Q−PAT(c)<−
with sIij=, where I{comparison statement} is 1 when the comparison statement holds and is 0 otherwise. The imprinting effect is classified into paternal imprinting effect if Q−PAT(c)<− , maternal imprinting effect if Q-PAT(c)>
, maternal imprinting effect if Q-PAT(c)> , and no imprinting effects otherwise. Here, α1 is the prespecified significance level for the first-stage imprinting test and
, and no imprinting effects otherwise. Here, α1 is the prespecified significance level for the first-stage imprinting test and  is the upper α1/2 quantile of a standard normal distribution. We then choose the optimal T2(c)-type test for association in the second stage based on the findings of Q-PAT(c). The following test is proposed to test for association:
is the upper α1/2 quantile of a standard normal distribution. We then choose the optimal T2(c)-type test for association in the second stage based on the findings of Q-PAT(c). The following test is proposed to test for association:

Alternatively, the maximum of the three T2(c)-type test statistics |T2m(c)|, |T2(c)| and |T2p(c)| is also considered, and we name the method as Q-MAX(c):

Test levels for Q-TDTI(c) and Q-MAX(c)
Under H0, T2m(c), T2(c), T2p(c) and Q-PAT(c) approximately follow a standard normal distribution, but they are not independent of each other. To obtain the significance levels of the Q-TDTI(c) and Q-MAX(c) test statistics, we need to derive the covariances involved under H0. As an example, the asymptotic covariance between T2(c) and Q-PAT(c) could be calculated by

where  , which is the unbiased estimate of the covariance between s2(c) and sI(c) under H0. The remaining covariances involved in the proposed tests could be similarly calculated (results omitted for brevity). Therefore, we obtain three 2 × 2 variance–covariance matrices for three bivariate normal distributions of T2m(c)/T2(c)/T2p(c) and Q-PAT(c) in Q-TDTI(c) and one 3 × 3 variance–covariance matrix for the trivariate normal distribution of T2m(c), T2(c) and T2p(c) in Q-MAX(c).
, which is the unbiased estimate of the covariance between s2(c) and sI(c) under H0. The remaining covariances involved in the proposed tests could be similarly calculated (results omitted for brevity). Therefore, we obtain three 2 × 2 variance–covariance matrices for three bivariate normal distributions of T2m(c)/T2(c)/T2p(c) and Q-PAT(c) in Q-TDTI(c) and one 3 × 3 variance–covariance matrix for the trivariate normal distribution of T2m(c), T2(c) and T2p(c) in Q-MAX(c).
Suppose that the overall significance level to test for association for both Q-TDTI(c) and Q-MAX(c) is fixed at α. Let α2 be the level that H0 is rejected in the second stage based on T2m(c), T2(c) or T2p(c). We have,

On the other hand, for the Q-MAX(c) test, let αm be the test level for each of the three tests that H0 is rejected, then we have,

Using the multivariate normal distribution, α2 and αm could be easily obtained by most statistical software; for example, R (http://www.r-project.org) and SAS Macro. Note that the constant c is included in Q-TDT(c) and Q-MAX(c). Although the validity of both methods does not depend on the value c, the corresponding power may be affected for different c values. To this end, as in Sun et al.,4 we choose c to be the mean trait value of all children in the sample.
Tests for both complete and incomplete nuclear families
Suppose that we have n1 incomplete nuclear families, which consist of nM families with only the maternal genotypes available (single-mother families) and nF families with only the paternal genotypes available (single-father families). The class of the T1(c) tests for association was suggested in Sun et al.4 The power of the T1(c) tests would be affected when there are imprinting effects. Therefore, on the basis of the tests 1−TDTp for the case of maternal imprinting and 1−TDTm for the case of paternal imprinting proposed by Xia et al.16 for association testing on qualitative trait loci, we develop two novel association tests T1p(c) and T1m(c). Specifically, T1p(c) and T1m(c) are constructed based on the nM single-mother families and the nF single-father families, respectively, which are expressed as  and
and  with
with


where  and
and  are analogously defined as
are analogously defined as  . Moreover, the class of T1(c) tests4 can be rewritten as
. Moreover, the class of T1(c) tests4 can be rewritten as  where s1(c)=s1p(c)+s1m(c) and
where s1(c)=s1p(c)+s1m(c) and  . It is shown in Supplementary Appendix I that T1p(c) and T1m(c) are valid for testing for association. On the basis of these three T1(c)-type test statistics T1p(c), T1(c) and T1m(c), we propose Q-1-TDTI(c) on the basis of the findings from the imprinting test Q-1-PAT(c)11 and Q-1-MAX(c), which are derived in the same framework as Q-TDTI(c) and Q-MAX(c), respectively.
. It is shown in Supplementary Appendix I that T1p(c) and T1m(c) are valid for testing for association. On the basis of these three T1(c)-type test statistics T1p(c), T1(c) and T1m(c), we propose Q-1-TDTI(c) on the basis of the findings from the imprinting test Q-1-PAT(c)11 and Q-1-MAX(c), which are derived in the same framework as Q-TDTI(c) and Q-MAX(c), respectively.
It is common in practice to collect a mixture of both complete and incomplete families. Suppose that we have n nuclear families under study, which are composed of n2 complete and n1 incomplete nuclear families. We propose the combined statistics to test for association, which are constructed through linear combinations of the corresponding T2(c)-type and T1(c)-type statistics. For instance,

Moreover, as in Q-TDTI(c) and Q-MAX(c), Q-C-TDTI(c) that employs Q-C-PAT(c)11 to test for imprinting, and Q-C-MAX(c) could be easily formulated for the combined data set. It is worth noting that the Q-C-TDTI(c) (Q-C-MAX(c)) is a versatile tool, and takes Q-TDTI(c) (Q-MAX(c)) and Q-1-TDTI(c) (Q-1-MAX(c)) as its special cases.
Simulation study
A simulation study is carried out to check the validity and to evaluate the performance of the proposed Q-C-TDTI(c) and Q-C-MAX(c) tests. We compare the proposed methods with the QTDT-G of Abecasis et al.3 and the QPL of Kistner et al.12 when the marker locus is in LD with the QTL and the marker locus is the QTL per se, respectively.
Size and power comparison with QTDT-G
We first study the size and powers of the proposed tests to test for association due to linkage disequilibrium. In the QTDT-G model, the trait value  for the jth child in the ith family is modeled as,
for the jth child in the ith family is modeled as,

where μ is the mean trait value, bi and wij are, respectively, the orthogonal between-family and within-family components of the genotype score of the individual, and  and
and  are similarly defined as bi and wij, but based on maternal transmission only. Under H0, the model is fitted with βw=0 and
are similarly defined as bi and wij, but based on maternal transmission only. Under H0, the model is fitted with βw=0 and  . For convenience, the identical by descent information required for QTDT-G are taken as the true values in our simulation study, thus the presented powers of QTDT-G are higher than what the test can actually achieve when the identical by descent information is estimated.
. For convenience, the identical by descent information required for QTDT-G are taken as the true values in our simulation study, thus the presented powers of QTDT-G are higher than what the test can actually achieve when the identical by descent information is estimated.
In simulating the family data sets, the population stratification model is considered by mixing two subpopulations in equal sampling proportions. The frequencies of maker allele M1 and allele D1 at the QTL are taken to be 0.3 (0.7) and 0.1 (0.5) in the first (second) subpopulation, respectively. Within each subpopulation, Hardy–Weinberg equilibrium is assumed. The recombination fraction between the ML and QTL is fixed at 0.001 in both subpopulations, and the LD coefficient between alleles M1 and D1 is measured by Lewinton’s D′.17 We use δ to denote the standardized LD coefficient in this paper. Let the mean trait values μ be 0 and 10 in the first and second subpopulations, respectively. The trait variance (σ2) is taken to be the sum of the variance of major-gene effect at the QTL  and the variance of environmental effect
and the variance of environmental effect  , which is taken as 100 in both subpopulations. Then, the trait values are generated from a normal distribution with mean trait value μ and variance σ2=100, though the normality assumption is not necessary for the proposed methods. Therefore, 20% of the total trait variance in the population under study is explained by the population stratification. Additive genetic models with different imprinting effects are considered in the simulation study.
, which is taken as 100 in both subpopulations. Then, the trait values are generated from a normal distribution with mean trait value μ and variance σ2=100, though the normality assumption is not necessary for the proposed methods. Therefore, 20% of the total trait variance in the population under study is explained by the population stratification. Additive genetic models with different imprinting effects are considered in the simulation study.
-
PI: model with paternal imprinting effect,
 ,
, 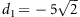 ,
, 
-
NI: model with no imprinting effects, a=10, d1=d2=0
-
MI: model with maternal imprinting effect,
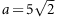 ,
,  ,
, 
 ,
, 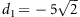 ,
, 
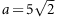 ,
,  ,
, 
This selection of the values of a, d1 and d2 leads to the heritablility ( /σ2) in the population being 18% in the first subpopulation and being 50% in the second subpopulation for all the three models.
/σ2) in the population being 18% in the first subpopulation and being 50% in the second subpopulation for all the three models.
Each simulated sample consists of 200 families each with one child and 200 families each with two children, so that the total number of children is 600 in each sample throughout the simulation study. To explore the influence of sample structure on the proposed tests, we define τ to be the ratio of incomplete nuclear families among all the families in each sample (termed as incomplete nuclear family ratio). Furthermore, we consider that the father or mother is missing at random in an incomplete nuclear family. All the simulation results are performed based on 1000 replicates and at the nominal 5% level.
The estimated type I error rates of the proposed tests (Q-C-TDTI(c) and Q-C-MAX(c)) and QTDT-G test are presented in Table 1. On the basis of 1000 replicates (with s.e. being 6.9 × 10−3), the estimates are in good agreement with the nominal 5% level regardless of imprinting effect and incomplete nuclear family ratio τ.
In the power study, we first compare the performance of Q-TDTI(c) and Q-MAX(c) with the existing QTDT-G method when there are only complete nuclear families (τ=0). Figure 1 plots the powers of Q-TDTI(c), Q-MAX(c), QTDT-G against standardized LD coefficient δ under the PI, NI and MI models, respectively. The corresponding powers for Q-PAT(c) of the Q-TDTI(c) approach are given in Table 2, from which we can see that the first stage Q-PAT(c) type test could effectively identify the imprinting effects, thus provides useful information on the selection of the appropriate association tests in the section stage. Overall, Q-TDTI(c) and Q-MAX(c) demonstrate superiority over the QTDT-G test for any level of δ. Q-TDTI(c) has slightly higher power than the proposed Q-MAX(c) test.

Simulated power estimates of Q-TDTI(c), Q-MAX(c) and QTDT-G against the standardized LD coefficient δ under PI, NI and MI models with incomplete family ratio τ=0, respectively. PI, model with paternal imprinting effect; NI, model with no imprinting effects; and MI, model with maternal imprinting effect.
Moreover, we compare the powers of the proposed tests and the QTDT-G test for different ratios of incomplete nuclear families. Figure 2 shows that the powers of the proposed tests Q-C-TDTI(c), Q-C-MAX(c) and the existing QTDT-G against the standardized LD coefficient δ for samples with τ being 0.25, 0.5 and 0.75, under the PI model. Note that the QTDT-G test can not handle this single parent case (τ=1). With the similar finding in Figure 1, both Q-C-TDTI(c) and Q-C-MAX(c) are more powerful than QTDT-G for any level of δ. The advantage is more significant with a greater proportion of incomplete nuclear families (with τ increasing), which is natural because QTDT-G only uses complete nuclear families in the sample. It is also noted that Q-C-TDTI(c) has slightly better performance than the proposed Q-C-MAX(c) test. Besides, we find that the performance of Q-C-TDTI(c) and Q-C-MAX(c) is better than QTDT-G under the MI and NI models (results omitted for brevity).

Simulated power estimates of Q-C-TDTI(c), Q-C-MAX(c) and QTDT-G against the standardized LD coefficient δ under PI model. Samples with different incomplete family ratios τ are considered (τ=0.25, 0.5 and 0.75). PI, model with paternal imprinting effect.
Size and power comparison with QPL
Further, we compare the performance of the proposed tests with the QPL test proposed by Kistner et al.12 The QPL test is constructed on quantitative polytomous logistic modeling, which can test for association incorporating imprinting effects. On the basis of the same simulation scenarios reported in Kistner et al.,12 we consider that the marker is the QTL per se. The population stratification model is used with the allele frequencies of M1 being 0.5 and 0.9 and the mean quantitative trait value being 0 and 1.5 in the first and second subpopulations, respectively. The quantitative trait is assumed to be normally distributed with a variance of 1.0 in both subpopulations. Further, the imprinting effects are simulated by imposing a shift λ, on the mean trait value for children inheriting a maternal copy of the allele. λ takes values from 0 to 2.4 in increments of 0.4. For all the scenarios considered, the simulated samples consist of either 300 or 500 parent–child trios. Furthermore, samples with a random 25% of the fathers missing have also been considered for assessing the powers of the tests in missing-parent cases. All the results of the proposed test are estimated based on 1000 replicates and at the nominal 5% level. Meanwhile, the results of QPL are taken from Kistner et al.12 for analyzing the same problem.
The results are plotted in Figure 3 and we can see that the proposed methods have similar accuracy with the QPL in controlling the size (λ=0). In power comparison, the proposed tests work much better than the QPL for all the scenarios simulated. The two proposed tests based on 300 families have even better performance than the QPL based on 500 families. Generally speaking, the proposed tests outperform QPL.

Simulated power estimates of Q-C-TDTI(c), Q-MAX(c) and QPL against imprinting shift λ. Samples with different sizes and missing father proportions are considered.
Application to FHS data
In this section, we apply the powerful and versatile approach Q-C-TDTI(c) to Framingham Heart Study data utilizing two traits: diastolic blood pressure and systolic blood pressure, and we are interested in detecting the genetic variants that may be associated with blood pressure. A few previous studies have revealed some genes associated with blood pressure, which show evidence in imprinting effects.11, 18 Therefore, we expect a gain in power for Q-C-TDTI(c) by incorporating the information on imprinting effects into association analysis.19 In the FHS data set, the sample came from three cohorts, which are the original cohort (the first generation), the offspring cohort (the second generation) and the gen3 group (the third generation). We select the independent nuclear families from the largest gen3 group that contains less missing genotype. The traits diastolic blood pressure and systolic blood pressure were measured at one time point in the gen3 group. After removing families with genotypes missing for both parents, we use the strategy of selecting family sample as follows: in each pedigree, if there exist complete nuclear families in its third generation, we select the largest complete nuclear family; otherwise, we select the largest incomplete nuclear family. Next, in each of the selected nuclear families, offspring without phenotype or genotype data (not informative for study) are removed. Finally, a total number of 592 nuclear families, a mixture of 322 complete nuclear families with both parents and 270 incomplete nuclear families with one parent, having 1327 children, enter into the sample for use in our application. On the basis of the phenotype and genotype of these 592 nuclear families, we totally scan 48 071 single-nucleotide polymorphism (SNP) markers on autosomes.
We fix the significance at 5% level for Q-C-PAT(c) in detecting imprinting effects in the first stage and at 1 × 10−4 level for testing association in the second stage, which are involved in Q-C-TDTI(c). Six SNPs, of which four for diastolic blood pressure and two for systolic blood pressure, are identified by Q-C-TDTI(c) to show the evidence for association with blood pressure. Their P-values are provided in Table 3. Among the six SNPs, the SNP rs2515663 is at the human blood pressure QTL 5 (BP5_H) from Rat Genome Database (URL: http://rgd.mcw.edu/) and the SNP rs12515112 and rs65802881 are at the human blood pressure QTL 21 (BP21_H). The remained three SNPs are at the locations where the rat blood pressure QTLs from rat genome database are mapped to Human. The SNP rs2515663 and rs6580288 have the most significant association with the blood pressure based on the proposed method.
Discussion
In this paper, we proposed two classes of association tests, Q-C-TDTI(c) and Q-C-MAX(c), by incorporating the information on imprinting effects into analysis. In our simulation study, we examined the validity and performance of the proposed tests. The simulation results demonstrated that the proposed methods controlled the size well under the null hypothesis of no association, and were more powerful than the existing QTDT-G method by Abecasis et al.4 and QPL method by Kistner et al.12 under the alternative when different levels of imprinting effects, different percents of missing data, etc., are considered. In addition, Q-C-TDTI(c) was found to be more powerful than Q-C-MAX(c). Finally, we successfully applied the powerful and versatile Q-C-TDTI(c) test to the Framingham Heart Study data set by using two blood pressure traits (diastolic blood pressure and systolic blood pressure) and showed the feasibility in practical application of the test.
Note that the proposed Q-C-TDTI(c) test has several promising advantages in application. First, by conditioning on the parental genotypes, Q-C-TDTI(c) is robust to population stratification. Second, Q-C-TDTI(c) requires no assumption on the distribution of the trait value. Apart from data sets based on random sampling, the test is also applicable to selectively genotyped samples; for example, individuals with trait values in the top 20% of the trait distribution are sampled (results are shown in Supplementary Appendix II). Third, compared with some other existing methods based on nuclear families with only one child, Q-C-TDTI(c) accommodates nuclear families with an arbitrary number of children and requires no identical by descent information for dealing with siblings data. Finally, Q-C-TDTI(c) is a versatile tool that can accommodate more complicated sample structures by incorporating incomplete nuclear family data into analysis and can also incorporate the effects of covariates, such as gender and age (refer to Supplementary Appendix III for details). The software for computing the proposed tests is currently available at http://lx2.saas.hku.hk/staff/wingfung/Q-TDTI/.
References
Spielman, R., McGinnis, R. & Ewens, W. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus. Am. J. Hum. Genet. 52, 506–516 (1993).
Allison, D. B. Transmission-disequilibrium tests for quantitative traits. Am. J. Hum. Genet. 60, 676–690 (1997).
Abecasis, G. R., Cardon, L. R. & Cookson, W. O. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 66, 279–292 (2000).
Sun, F. Z., Flanders, W. D., Yang, Q. H. & Zhao, H. Y. Transmission/disequilibrium tests for quantitative traits. Ann. Hum. Genet. 64, 555–565 (2000).
Van den Oord, E. J. The use of mixture models to perform quantitative tests for linkage disequilibrium, maternal effects, and parent-of-origin effects with incomplete subject-parent triads. Behav. Genet. 30, 335–343 (2000).
Li, J. M., Wang, D., Dong, J. P., Jiang, R. F., Zhang, K., Zhang, S. L. et al. The power of transmission disequilibrium tests for quantitative traits. Genet. Epidemiol. 21, S632–S637 (2001).
Gauderman, W. J. Candidate gene association analysis for a quantitative trait, using parent-offspring trios. Genet. Epidemiol. 25, 327–338 (2003).
Kistner, E. O. & Weinberg, C. R. Method for using complete and incomplete trios to identify genes related to a quantitative trait. Genet. Epidemiol. 27, 33–42 (2004).
Kistner, E. O. & Weinberg, C. R. A method for identifying genes related to a quantitative trait, incorporating multiple siblings and missing parents. Genet. Epidemiol. 29, 155–165 (2005).
Wheeler, E. & Cordell, H. J. Quantitative trait association in parent offspring trios: extension of case/pseudocontrol method and comparison of prospective and retrospective approaches. Genet. Epidemiol. 31, 813–833 (2007).
He, F., Zhou, J. Y., Hu, Y. Q., Sun, F., Yang, J., Lin, S. et al. Detection of parent-of-origin effects for quantitative traits in complete and incomplete nuclear families with multiple children. Am. J. Epidemiol. 174, 226–233 (2011).
Kistner, E. O., Infante-Rivard, C. & Weinberg, C. R. A method for using incomplete triads to test maternally mediated genetic effects and parent-of-origin effects in relation to a quantitative trait. Am. J. Epidemiol. 163, 255–261 (2006).
Belonogova, N. M., Axenovich, T. I. & Aulchenko, Y. S. A powerful genome-wide feasible approach to detect parent-of-origin effects in studies of quantitative traits. Eur. J. Hum. Genet. 18, 379–384 (2010).
Feng, R., Wu, Y., Jang, G. H., Ordovas, J. M. & Arnett, D. A powerful test of parent-of-origin effects for quantitative traits using haplotypes. PLoS ONE 6, e28909 (2011).
Hu, Y. Q., Zhou, J. Y. & Fung, W. K. An extension of the transmission disequilibrium test incorporating imprinting. Genetics 175, 1489–1504 (2007).
Xia, F., Zhou, J. Y. & Fung, W. K. A powerful approach for association analysis incorporating imprinting effects. Bioinformatics 27, 2571–2577 (2011).
Lewontin, R. On measures of gametic disequilibrium. Genetics 120, 849–852 (1988).
Yang, J. & Lin, S. Detection of imprinting and heterogeneous maternal effects on high blood pressure using Framingham Heart Study data. BMC Proc. 3, S125 (2009).
Adeyemo, A., Gerry, N., Chen, G., Herbert, A., Doumatey, A. et al. A genome-wide association study of hypertension and blood pressure in african Americans. PLoS Genet. 5, e1000564 (2009).
Acknowledgements
We thank the reviewers for valuable comments that largely improve the presentation of the manuscript. This work was supported by the Hong Kong RGC Research Grant (766511M), the National Natural Science Foundation of China (81072386) and grants from School of Public Health and Tropical Medicine of Southern Medical University, China (grant numbers: GW201219 and GW201237). The Framingham Heart Study research was supported by NHLBI contract: 2 N01-HC-25195-06 and its contract with Affymetrix Inc for genotyping services (contract no. N02-HL-6-4278). The Genetic Analysis Workshop is supported by NIH grant R01 GM31575. The GAW16 Framingham data used for the analyses described in this paper were obtained through dbGaP (no. 7325-3 and no. 7640-2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Xia, F., Zhou, JY. & Fung, W. Powerful tests for association on quantitative trait loci incorporating imprinting effects. J Hum Genet 58, 384–390 (2013). https://doi.org/10.1038/jhg.2013.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.22
Keywords
This article is cited by
-
Robust association tests for quantitative traits on the X chromosome
Heredity (2022)
-
A powerful association test for qualitative traits incorporating imprinting effects using general pedigree data
Journal of Human Genetics (2015)
