
Abstract
The events that take place during the prophase of meiosis I are essential for the correct segregation of homologous chromosomes. Defects in these processes likely contribute to infertility or recurrent pregnancy loss in humans. To screen for candidate genes for reproductive failure due to meiotic defects, we have analyzed the gene expression patterns in fetal, neonatal and adult gonads of both male and female mice by microarray and thereby identified 241 genes that are expressed specifically during prophase of meiosis I. Combined with our previous data obtained from developing spermatocytes, a total of 99 genes were identified that are upregulated in early prophase I. We confirmed the meiotic prophase I-specific expression of these genes using qRT-PCR. To further screen this panel for candidate genes that fulfill important roles in homologous pairing, synapsis and recombination, we established a gene transfer system for prophase I oocytes in combination with in vitro organ culture of ovaries, and successfully determined the localization of the selected genes. This gene set can thus serve as a resource for targeted sequence analysis via next-generation sequencing to identify the genes associated with human reproduction failure due to meiotic defects.
Similar content being viewed by others


Introduction
Infertility is a serious clinical condition for couples who desire to have children. It is estimated that ∼15% of couples attempting conception suffer from some form of infertility, and that in ∼30% of these cases there is no definitive cause.1 Although some infertile couples are successfully treated with assisted reproductive technology, some couples can go through years of infertility treatment without success. Another serious problem in reproduction is recurrent pregnancy loss (RPL), which affects ∼5% of couples.2 The cause of RPL cannot be determined in up to 50% of the cases.3, 4 As multiple factors underlie the etiology of infertility and RPL, these disorders are believed to be polygenic, whereas single-gene abnormalities have been demonstrated to be a cause of reproductive defects in animal models.5 The heterogeneous and complex characteristics of RPL have made the causes challenging to elucidate. Moreover, it is difficult to identify the responsible gene(s) using pedigree analysis because the genetic factors that cause a susceptibility to infertility will be poorly transmitted through the germline.
Mutations or polymorphisms in genes associated with meiosis are potentially good candidates as genetic causes of infertility because they would create no other defects other than reproductive defects. Approaches involving the sequencing of candidate genes have enabled the identification of a few meiotic genes that are causal for infertility or RPL.6, 7, 8, 9 Further analyses of these candidate genes will be necessary to better understand the genetic basis of human infertility and RPL. It is likely also that many other genes involved in meiosis in mammals, and that that remain to be identified, have a role in these disorders.
Unlike mitotic division, homologous chromosomes segregate from each other during meiosis I. The formation of DNA double-strand breaks, subsequent repair by homologous recombination and maintenance of the resultant chiasma between homologous chromosomes until anaphase are essential for correct segregation. Studies in mouse models have revealed that impairment of these processes during early meiosis results in apoptosis or aneuploidy.10 The extensive apoptosis of gametes leads to infertility, whereas aneuploidy in gametes, whether the abnormality is derived from a homologous or sister chromosome segregation error, contributes to RPL or to the birth of offspring with congenital defects. Interestingly, males and females differ in terms of meiotic defect outcomes. In males, infertility due to extensive apoptosis is the most predominant outcome, whereas segregation errors in maternal chromosomes often results in aneuploid oocytes.11 It has been reasonably hypothesized that this sexual dimorphism might be due to a prolonged meiotic prophase I in females or due to differences in checkpoint robustness.12, 13 However, despite its importance to understanding the differences in the molecular mechanisms underlying correct segregation between males and females, there is little currently known about oogenesis because of the limited availability of oocytes.
To further investigate the etiology of infertility and RPL in humans, and the mechanisms of chromosome segregation in mammals, we have previously performed expression profiling analysis of mouse spermatocytes.14 We focused on screening for genes upregulated in the transition to leptotene/zygotene spermatocytes from spermatogonia to identify the factors controlling the correct segregation of chromosomes during meiosis I. We thereby identified a total of 726 genes, MLZ (male leptotene and zygotene)-001-726. However, this MLZ gene panel actually included many spermatogenesis-specific genes, which hindered further analyses. In our current study, the gene expression profiles of both male and female gonads undergoing meiosis I were examined by microarray analysis to exclude the effects of male-specific genes in the MLZ gene set. This enabled an enrichment of meiotic genes and prevented the exclusion of female factors.
Materials and methods
Microarray analysis
All animal experiments were approved by the Animal Care and Use Committee at Fujita Health University. Total RNA was extracted from male and female gonads of C57BL6 mice at 15.5 days postcoitum (dpc), neonatal day 1 and 10 weeks postpartum. Microarray experiments were performed as described previously,14 except that the GeneChip Mouse Genome 430 2.0 array (Affymetrix, Santa Clara, CA, USA) was used in this study. Microarray data analyses were performed using Microsoft Excel (Microsoft, Redmond, WA, USA), Genespring software, version 11 (Silicon Genetics, Redwood City, CA, USA) and Affymetrix Microarray Suite, Version 5 (MAS5, Affymetrix). All data were subjected to per chip and per gene normalization, and then used for further analysis. Annotations of all filtered transcripts were updated using Affymetrix NetAffx (http://www.netaffx.com), based on the July 2011 annotation update. Microarray data were deposited in the GEO database according to the MIAME Guidelines and assigned the accession number GSE35734.
Real-time RT-PCR
First-strand cDNAs were synthesized as described previously.14 Real-time RT-PCR assays were also performed as described previously.15 The primer sets used in these experiments are listed in Supplementary Table 1.
Expression of epitope-tagged proteins
The entire coding region of each selected candidate gene was obtained by RT-PCR and individually cloned into the pFLAG-CMV2 vector (Sigma-Aldrich, St Louis, MO, USA). Plasmids were dissolved in electroporation buffer (125 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 10 mM glucose and 20 mM Hepes, pH 7.4)16 at a concentration of 3.5 μg μl−1. Mouse ovaries with attached mesonephroses at 13.5 dpc were removed. After injection of the tissues with plasmids suspended in L-15 medium, the ovaries were electroporated as described previously17 with some modifications. Briefly, after injection, ovaries with mesonephroses were placed between electrodes (CUY501G2, Nepa Gene, Chiba, Japan) with a small volume of electroporation buffer. A 45-V, 50-ms rectangular pulse was charged 5 times at 100-ms intervals using an electroporator (CUY21SC, Nepa Gene). The tissues were then cultured for 2 days as described previously17 except that Transwell membrane inserts (polycarbonate membrane, 24-mm diameter, 3.0-μm pore size, Corning, Corning, NY, USA) and minimum essential medium-alpha medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum, and supplemented with penicillin and streptomycin were used. For expression in mouse spermatocytes, the plasmids were introduced into the testis by in vivo electroporation as described previously,9 and these tissues were dissected out for immunostaining 2 days later.
Immunofluorescence microscopy
Testis frozen sections were fixed in acetone for 15 min and then washed three times in phosphate-buffered saline (PBS). After blocking with 10% normal donkey serum in PBS for 30 min at room temperature, primary antibodies were applied as described below. Squashed oocytes were prepared as described previously.18, 19 Slides were incubated with blocking buffer (5% normal donkey serum and 5% normal goat serum in PBS), and then incubated with primary antibodies diluted in PBS at 4 °C overnight at the following dilutions: rabbit anti-FLAG (D-8, Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1:1000; guinea-pig anti-SYCP3 antiserum,15 1:5000. The slides were then washed three times in PBS for 15 min, and incubated with the following secondary antibodies to enable detection by fluorescence microscopy: Alexa 594-conjugated donkey anti-rabbit IgG and Alexa 488-conjugated goat anti-guinea-pig IgG (Invitrogen), or aminomethylcoumarin acetate-conjugated donkey anti-guinea-pig IgG (Jackson ImmunoReasearch, West Grove, PA, USA). The secondary antibodies were diluted in PBS and incubated at room temperature for 30 min. After again washing three times in PBS, slides were mounted in Mowiol/DABCO (Sigma-Aldrich) with or without DAPI (4′-6-diamidino-2-phenylindole) (0.5 μg ml−1) and observed under a fluorescence microscope (Axio Imager M1, Carl Zeiss, Jena, Germany). Images were processed using Adobe Photoshop software (Adobe Systems, San Jose, CA, USA).
RESULTS
Enrichment of a candidate gene set for meiosis I prophase in mouse gonads
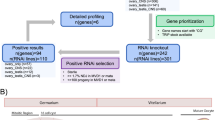
The gene expression profiles of gonads from a 15.5-dpc fetal male (FM), 15.5-dpc fetal female (FF), neonatal day 1 male (NM), neonatal day 1 female (NF), 10-week-old male (AM) and 10-week-old female (AF) mice were examined by microarray analysis. To screen for meiotic prophase I-related genes, we performed filtering as described in Figure 1a. First, we eliminated male-specific genes using flag calls as follows. Genes with a ‘present’ or ‘marginal’ call in either FF or NF were selected, which would be expected to include genes expressed in female meiosis I but no male-specific genes. Likewise genes with a ‘present’ or ‘marginal’ call in AM were selected to eliminate female-specific genes. These genes were further filtered by comparing the expression levels in AM with those in FM, NM and AF, followed by filtering of genes for which the expression levels in FF were more than threefold higher than those of AF. Thus, we extracted a total of 267 genes that were upregulated in both male and female gonads undergoing meiotic prophase I. After correcting for redundancy, 241 non-redundant genes were identified and these are listed in Supplementary Table 2. Within this panel, there were 99 matches for genes in the MLZ gene set (Figure 1b), and these are listed in Supplementary Table 3.

Generation of a gene set that is upregulated during meiotic prophase I. (a) Strategy for filtering genes that are upregulated in the mouse gonads undergoing meiotic prophase I. ‘Present’ and ‘Marginal’ are detection calls of the MAS5 algorithm, which is an indicator of expression. The numbers of genes selected by each step are indicated. (b) Venn diagram indicating the number of selected genes using two criteria: the MLZ gene set identified previously by Kogo et al.14 (left) and genes identified in the current analysis (right).
The composition of the final 99 selected genes is summarized in Table 1. Among these gene candidates, 76 are named and the remaining 23 are previously uncharacterized. Among the named genes, 17 genes (∼22%) have been reported to show meiosis-preferred expression, with only six genes known to display testis-preferred expression. On the other hand, among the 578 named genes in the MLZ gene set identified previously, 101 genes have been demonstrated to show testis-preferred expression.14 This indicates that the male-specific genes had mostly been excluded from the MLZ gene set in this study. Significantly, most of the genes known to function in meiotic prophase I were included in our final 99 gene panel, as shown in Table 2. These data suggest that the MLZ gene set had been successfully enriched with genes involved in the prophase of meiosis I. However, three genes, Mei1, Msh5 and Rec8, in spite of their well-known functional association with chromosomal behavior in meiosis, did not appear among these 99 genes, but were included in the initial screen of 241 genes. We thus performed subsequent analyses also on this larger 241 gene set.
Developmental gene expression profiling of the female mouse gonads
To explore which of the candidate genes in our data sets show a similar expression profile with known meiotic genes, we grouped the 241 and 99 gene panels discretely into 4 clusters using k-means clustering analysis. Three groups of microarray data from mouse female gonads were applied to this analysis in order to focus on the expression level changes between the early (15.5 dpc) and late (neonatal day 1) stages of prophase I. The results of our clustering of the 241 genes are shown in Figure 2a and Supplementary Table 2. For the genes in cluster A, the expression levels are higher in NF than in FF or AF. Fifty genes were grouped in cluster A, but none with known functions in meiotic chromosomal behavior and only 11 that overlapped with the 99 selected meiotic genes. Hence, cluster A appeared to contain female-predominant genes expressed in the later stages of oogenesis. Cluster B contained as many as 110 genes, but only five with meiosis-specific expression, suggesting that many of the genes in this cluster may not be specifically associated with meiotic chromosomal behavior. The gene expression patterns for cluster C were found to be similar to those of cluster B, although the expression levels in FF were higher than the genes in cluster B. Cluster C contains 10 meiotic genes including Spo11, Rec8 and Sycp3. Genes in cluster D exhibited the highest expression levels in FF compared with the other clusters. Among the 27 genes in this cluster, 22 overlapped with the 99 gene panel, and five of these genes are well-known meiotic genes including Dmc1 and Smc1b. These results confirm the enrichment of genes involved in meiosis I prophase in the 99 gene set.

K-means clustering for the gene expression profiles of the 241 and 99 gene panels derived from the developmental mouse ovary. (a) Clustering of the 241 gene panel. Numbers indicate the gene numbers of the 241 panel, whereas those in parentheses denote the candidates in the 99 gene panel. (b) Clustering of the 99 gene panel. Numbers indicate the gene numbers of this panel. Normalized signal intensities are shown on the y axis. Because some genes have more than one probe set on the microarray platform, the sum of the four clusters exceeds 241 or 99 (see Supplementary Tables 2 and 3).
Similar clustering was also performed for the 99 selected genes (Figure 2b and Supplementary Table 3). The expression levels of the genes assigned to cluster 1 were high in FF but decreased in NF. Spo11, Dmc1 and Atm, which are the key enzymes involved in the formation and repair of meiotic DNA double-strand breaks, were included in this cluster. The expression levels of the genes in cluster 2 were higher in both FF and NF than those of the cluster 1 genes. This cluster includes a cohesin component, Smc1b. Hence, clusters 1 and 2 may include genes involved in early stages of prophase during meiosis I. Cluster 3 contains genes whose expression did not switch off until neonatal day 1. Sycp1, Sycp3 and Syce1 are included in this cluster, which indicates that the genes in cluster 3 are necessary through to the later stages of prophase I. The genes in cluster 4 showed lower expression in both FF and NF than the genes of any other clusters. Syce2 is the only gene in this cluster to have been functionally established as a meiotic gene, indicating that our 99 gene panel still included non-meiotic genes that were categorized into cluster 4.
Real-time RT-PCR analysis of selected genes
We randomly selected nine genes from the 99 gene panel with no known function in meiosis (Table 3). These genes were analyzed by real-time RT-PCR to verify whether they were meiosis-specific (Figure 3). We used RNA extracts from the gonads of FM, FF, AM and AF mice, because we were focusing only on the early stages of prophase I. The expression of Sycp3, a meiosis-specific gene, showed high levels in FF and AM, but low levels in FM, AF and somatic tissues (Figure 3a). MLZ-175, MLZ-611, MLZ-638 and MLZ-675 showed meiotic expression patterns that were similar to Sycp3 (Figure 3a). MLZ-254, MLZ-326 and MLZ-344 showed testis-predominant expression patterns (Figure 3b). MLZ-352 and MLZ-617 showed testis-predominant expression patterns, but were found to be expressed also in the adult mouse ovary (Figure 3b). In 11 of the known meiotic genes listed in Supplementary Table 3, the expression ratios of FF to AF derived from the microarray analysis were high (>22-fold). These genes ranked in the top 25 among the 99 gene panel in terms of the ratios of FF to AF. Among the nine genes we analyzed by real-time RT-PCR, MLZ-611 and MLZ-675 ranked also in the top 25, and both showed meiotic expression patterns (Figure 3a). These results suggest that genes with a higher expression ratio of FF to AF among our 99 selected genes are strong candidates as functional genes in meiotic chromosomal behavior.

Real-time RT-PCR analysis of selected candidate genes using RNA from fetal gonads obtained from male and female mice at 15.5 dpc; adult gonads obtained from male and female mice at 10 weeks postpartum and somatic tissues. (a) Genes exhibiting meiotic expression patterns. (b) Genes exhibiting testis-predominant expression patterns. Expression ratios relative to the FM are shown on the y axis. Samples were run in triplicate, and error bars represent the standard deviation.
Subcellular localization of the epitope-tagged proteins expressed in mouse oocytes
To perform a further screen for meiotic genes that might be involved in chromosome segregation, we examined the subcellular localization of the protein products encoded by our candidate genes using vectors expressing epitope-tagged proteins. In our previous study, HEK293 cells had been used as a heterologous transfection system.14 However, ectopically expressed SYCP3 generates a loop-like structure not only in the nucleus but also in the cytoplasm in somatic cell lines, which had not correctly reflected its intrinsic localization pattern in a previous report.9 Therefore, in our current analyses we introduced a plasmid DNA expressing FLAG-tagged SYCP3 into adult mouse testis by in vivo electroporation. The FLAG-tagged SYCP3 protein product was detectable in the nuclei of pachytene spermatocytes by immunofluorescence with anti-FLAG antibodies, and the staining pattern was found to be similar to that of endogenous SYCP3, reflecting that it is a component of the synaptonemal complex (Figure 4a). As we were focusing on the genes associated with female meiosis in the analysis, the same plasmid was introduced into a fetal mouse ovary by electroporation followed by in vitro organ culture. Overexpressed SYCP3 with a FLAG-tag was found to localize in the nuclei of leptotene oocytes as also demonstrated in spermatocytes (Figure 4b). These results indicate that epitope-tagged proteins that are overexpressed in both spermatocytes and oocytes show a pattern that reflects their intrinsic subcellular localization during meiosis. We thus applied this method to oocytes to examine the subcellular localization of unknown genes.

Subcellular localization of candidate meiotic gene protein products. Plasmids expressing FLAG-tagged proteins were introduced into mouse testis or ovarian tissues. (a) Immunostaining of testis frozen sections using anti-FLAG antibodies (red) and anti-SYCP3 antiserum (blue). Bar, 10 μm. (b) Squashed oocytes were analyzed by immunostaining with anti-FLAG antibodies (red), anti-SYCP3 antiserum (green), and nuclear counterstaining was performed with DAPI (blue). Nu, nucleus; Cy, cytoplasm. Bar, 20 μm.
Plasmids expressing FLAG-tagged protein products for two candidate genes (MLZ-611 and MLZ-638) were introduced into oocytes (Figure 4b). The amino acid sequences encoded by these genes had no obvious nuclear localization signals and no distinct motifs other than coiled-coils. The protein encoded by MLZ-638 was observed in the cytoplasm of the oocytes. The localization of the MLZ-611 protein showed two patterns: both nuclear and cytoplasmic staining, and cytoplasmic only. The nuclear localization of MLZ-611 suggests a possible role in chromosome segregation.
Discussion
To date, studies of global gene expression profiles of isolated oocytes have been mostly limited to metaphase II oocytes or earlier oocytes at the primordial follicle stage.20, 21, 22, 23 Although gene expression profile data for fetal or neonatal ovaries have been provided in mouse and human, and have enabled the analysis of genes involved in the initiation of meiosis, the sex differentiation of the gonad, early meiosis, and ovarian and follicle development, investigations of meiotic prophase I have been lacking.24, 25, 26, 27, 28 In yeast, it is estimated that ∼150 genes may be meiosis-specific.29, 30 In our current study, we screened 99 genes that are upregulated during prophase in meiosis I. Despite the fact that this gene number is less than one-seventh of the previous MLZ gene set, most of the well-known genes associated with meiotic chromosome behavior, sister chromatid cohesion, synapsis and homologous recombination, are included in this select gene panel. This indicates that our select 99 gene panel includes many other unknown meiotic genes, the functional elucidation of which will likely facilitate an increased understanding of the mechanisms underlying the sexual dimorphism between males and females. For further screening, 76 uncharacterized genes among these 99 genes is a reasonable number for individual validation by qRT-PCR. In addition, genes showing high FF to AF expression ratios by microarray should have priority in this respect.
Expression profiling using k-means clustering revealed that well-established meiotic genes were grouped into the same clusters, depending on their functions. Our clustering analysis of the initial 241 candidate gene panel showed that most of the known meiotic genes in this set grouped into two clusters, C and D. Hence, genes in these clusters that did not overlap with the 99 gene series may include genes that are upregulated after the zygotene stage and therefore warrant further analysis. Cluster 2, exhibiting the highest expression level in FF and NF among the four clusters established for the smaller 99 gene panel, included only one functionally known meiotic gene, Smc1b. This gene is important for maintaining the chiasma until adulthood, although its expression is clearly detectable only during the fetal stages and is barely evident in later stages in the female.31 As the loading of SMC1B onto chromosomes begins at the leptotene stage during prophase I to establish meiotic cohesion, this protein needs to remain functional during a long meiotic arrest period in which it is not replenished.19, 32 The high expression of Smc1b transcripts in NF mice implies that robust cohesion might be established by this stage. A lack of turnover of the meiotic cohesin protein is considered to contribute to age-related chromosome segregation errors in oocytes.32, 33, 34 The molecular mechanisms that maintain or disrupt meiotic cohesion have remained unclear for many decades. However, the candidate genes in cluster 2 in our current study may assist with our understanding of this complex phenomenon going forward.
Transient expression assays of epitope-tagged proteins to examine their subcellular localizations in oocytes would also likely assist with a determination of whether the molecules are engaged in chromosome behavior by direct observation of nuclear localization. The possible effect of the expression levels to the resultant subcellular localization might be overcome by observation of cells with various expression levels. As the efficiency of gene transfer for each gene varied from 2–3 cells to 20–30 cells per a litter, optimization of the condition for introduction might be necessary for each gene. To our knowledge, this is the first report to screen for genes of interest and then functionally characterize some of these candidates via their introduction into mouse oocytes during prophase I. Using this gene transfer system, the functions of uncharacterized gene can begin to be predicted if the localization of their encoded proteins is a unique nuclear domain, for example, at the centromeres or chiasmata on meiotic chromosomes. Further, when a mutation has been identified in individuals with infertility or RPL, introduction of the mutant gene into oocytes in this way would readily enable functional analysis without the need to establish a knock-in mouse.
Also in our current analysis, we have identified that an uncharacterized gene, MLZ-611, may function in homologous chromosome segregation on the basis of its localization at the nucleus during prophase I. Recently, a bioinformatics approach using data from the published microarray analyses of gene expression during prophase I has provided a cohort of candidate meiotic genes.35 The authors of this report performed expression profiles of male and female gonads from mouse and human, and compared these patterns to detect conserved co-expression networks. Interestingly, MLZ-611 has a co-expression link with Sycp3, and MLZ-638 has co-expression links with Smc1b, Sycp3, Hormad2, and Dmc1 across mouse and human systems. This indicates that MLZ-611 and MLZ-638 have potential functional connections with well-known meiotic genes in human.
Our final goal in the present study was to identify genes that may have a role in human infertility or RPL. Genes in our candidate lists that are verified as meiotic genes will be further assessed in the future using high-throughput mutational analysis involving targeted next-generation sequencing of genomic DNA from patients with reproductive problems. Recently, exome sequencing has been widely applied to the identification of causative mutations for human diseases.36, 37, 38 However, this approach has been mainly limited to analyses of rare diseases because it is still too costly at present, to screen for mutations responsible for common diseases such as infertility or RPL in this way. Reducing the number of genes to be analyzed, as well as employing simultaneous analysis of multiple samples using barcoded primers assigned to each patient,39 will allow the genome analyses to be conducted more cost-effectively and more rapidly.
Accession codes
References
Smith, S., Pfeifer, S. M., Collins, J. A. Diagnosis and management of female infertility. JAMA 290, 1767–1770 (2003).
Sierra, S., Stephenson, M. Genetics of recurrent pregnancy loss. Semin. Reprod. Med. 24, 17–24 (2006).
Stephenson, M. D. Frequency of factors associated with habitual abortion in 197 couples. Fertil. Steril. 66, 24–29 (1996).
Li, T. C., Makris, M., Tomsu, M., Tuckerman, E., Laird, S. Recurrent miscarriage: aetiology, management and prognosis. Hum. Reprod. Update 8, 463–481 (2002).
Matzuk, M. M., Lamb, D. J. The biology of infertility: research advances and clinical challenges. Nat. Med. 14, 1197–1213 (2008).
Miyamoto, T., Hasuike, S., Yogev, L., Maduro, M. R., Ishikawa, M., Westphal, H. et al. Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet 362, 1714–1719 (2003).
Christensen, G. L., Ivanov, I. P., Atkins, J. F., Mielnik, A., Schlegel, P. N., Carrell, D. T. Screening the SPO11 and EIF5A2 genes in a population of infertile men. Fertil. Steril. 84, 758–760 (2005).
Mandon-Pépin, B., Touraine, P., Kuttenn, F., Derbois, C., Rouxel, A., Matsuda, F. et al. Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur. J. Endocrinol. 158, 107–115 (2008).
Bolor, H., Mori, T., Nishiyama, S., Ito, Y., Hosoba, E., Inagaki, H. et al. Mutations of the SYCP3 gene in women with recurrent pregnancy loss. Am. J. Hum. Genet. 84, 14–20 (2009).
Cohen, P. E., Pollack, S. E., Pollard, J. W. Genetic analysis of chromosome pairing, recombination, and cell cycle control during first meiotic prophase in mammals. Endocr. Rev. 27, 398–426 (2006).
Hassold, T., Hall, H., Hunt, P. The origin of human aneuploidy: where we have been, where we are going. Hum. Mol. Genet. 16, R203–R208 (2007).
Kurahashi, H., Bolor, H., Kato, T., Kogo, H., Tsutsumi, M., Inagaki, H. et al. Recent advance in our understanding of the molecular nature of chromosomal abnormalities. J. Hum. Genet. 54, 253–260 (2009).
Kogo, H., Tsutsumi, M., Ohye, T., Inagaki, H., Abe, T., Kurahashi, H. HORMAD1-dependent checkpoint/surveillance mechanism eliminates asynaptic oocytes. Genes Cells 17, 439–454 (2012).
Kogo, H., Kowa-Sugiyama, H., Yamada, K., Bolor, H., Tsutsumi, M., Ohye, T. et al. Screening of genes involved in chromosome segregation during meiosis I: toward the identification of genes responsible for infertility in humans. J. Hum. Genet. 55, 293–299 (2010).
Tsutsumi, M., Kogo, H., Kowa-Sugiyama, H., Inagaki, H., Ohye, T., Kurahashi, H. Characterization of a novel mouse gene encoding an SYCP3-like protein that relocalizes from the XY body to the nucleolus during prophase of male meiosis I. Biol. Reprod 85, 165–171 (2011).
Kawabata, I., Umeda, T., Yamamoto, K., Okabe, S. Electroporation-mediated gene transfer system applied to cultured CNS neurons. Neuroreport 15, 971–975 (2004).
Nakamura, Y., Yamamoto, M., Matsui, Y. Introduction and expression of foreign genes in cultured mouse embryonic gonads by electroporation. Reprod. Fertil. Dev. 14, 259–265 (2002).
Page, J., Suja, J. A., Santos, J. L., Rufas, J. S. Squash procedure for protein immunolocalization in meiotic cells. Chromosome Res. 6, 639–642 (1998).
Prieto, I., Tease, C., Pezzi, N., Buesa, J. M., Ortega, S., Kremer, L. et al. Cohesin component dynamics during meiotic prophase I in mammalian oocytes. Chromosome Res. 12, 197–213 (2004).
Hamatani, T., Falco, G., Carter, M. G., Akutsu, H., Stagg, C. A., Sharov, A. A. et al. Age-associated alteration of gene expression patterns in mouse oocytes. Hum. Mol. Genet. 13, 2263–2278 (2004).
Pan, H., O’brien, M. J., Wigglesworth, K., Eppig, J. J., Schultz, R. M. Transcript profiling during mouse oocyte development and the effect of gonadotropin priming and development in vitro. Dev. Biol. 286, 493–506 (2005).
Kocabas, A. M., Crosby, J., Ross, P. J., Otu, H. H., Beyhan, Z., Can, H. et al. The transcriptome of human oocytes. Proc. Natl Acad. Sci. USA 103, 14027–14032 (2006).
Grøndahl, M. L., Yding Andersen, C., Bogstad, J., Nielsen, F. C., Meinertz, H., Borup, R. Gene expression profiles of single human mature oocytes in relation to age. Hum. Reprod. 25, 957–968 (2010).
Small, C. L., Shima, J. E., Uzumcu, M., Skinner, M. K., Griswold, M. D. Profiling gene expression during the differentiation and development of the murine embryonic gonad. Biol. Reprod. 72, 492–501 (2005).
Gallardo, T. D., John, G. B., Shirley, L., Contreras, C. M., Akbay, E. A., Haynie, J. M. et al. Genomewide discovery and classification of candidate ovarian fertility genes in the mouse. Genetics 177, 179–194 (2007).
Olesen, C., Nyeng, P., Kalisz, M., Jensen, T. H., Møller, M., Tommerup, N. et al. Global gene expression analysis in fetal mouse ovaries with and without meiosis and comparison of selected genes with meiosis in the testis. Cell. Tissue. Res. 328, 207–221 (2007).
Houmard, B., Small, C., Yang, L., Naluai-Cecchini, T., Cheng, E., Hassold, T. et al. Global gene expression in the human fetal testis and ovary. Biol. Reprod. 81, 438–443 (2009).
Hogarth, C. A., Mitchell, D., Evanoff, R., Small, C., Griswold, M. Identification and expression of potential regulators of the mammalian mitotic-to-meiotic transition. Biol. Reprod. 84, 34–42 (2011).
Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P. O. et al. The transcriptional program of sporulation in budding yeast. Science 282, 699–705 (1998).
Primig, M., Williams, R. M., Winzeler, E. A., Tevzadze, G. G., Conway, A. R., Hwang, S. Y. et al. The core meiotic transcriptome in budding yeasts. Nat. Genet. 26, 415–423 (2000).
Hodges, C. A., Revenkova, E., Jessberger, R., Hassold, T. J., Hunt, P. A. SMC1beta-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat. Genet. 37, 1351–1355 (2005).
Revenkova, E., Herrmann, K., Adelfalk, C., Jessberger, R. Oocyte cohesin expression restricted to predictyate stages provides full fertility and prevents aneuploidy. Curr. Biol. 20, 1529–1533 (2010).
Tachibana-Konwalski, K., Godwin, J., van der Weyden, L., Champion, L., Kudo, N. R., Adams, D. J. et al. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev. 24, 2505–2516 (2010).
Chiang, T., Schultz, R. M., Lampson, M. A. Meiotic origins of maternal age-related aneuploidy. Biol. Reprod. 86, 1–7 (2012).
Su, Y., Li, Y., Ye, P. Mammalian meiosis is more conserved by sex than by species: conserved co-expression networks of meiotic prophase. Reproduction 142, 675–687 (2011).
Bamshad, M. J., Ng, S. B., Bigham, A. W., Tabor, H. K., Emond, M. J., Nickerson, D. A. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755 (2011).
Ku, C. S., Naidoo, N., Pawitan, Y. Revisiting Mendelian disorders through exome sequencing. Hum. Genet. 129, 351–370 (2011).
Singleton, A. B. Exome sequencing: a transformative technology. Lancet Neurol. 10, 942–946 (2011).
Parameswaran, P., Jalili, R., Tao, L., Shokralla, S., Gharizadeh, B., Ronaghi, M. et al. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 35, e130 (2007).
Acknowledgements
We thank Dr Hiroki Kano and Ms Chiaki Shimizu for technical assistance. These studies were supported by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and a grant from the Hori Information Science Promotion Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Tsutsumi, M., Kowa-Sugiyama, H., Bolor, H. et al. Screening of genes involved in chromosome segregation during meiosis I: in vitro gene transfer to mouse fetal oocytes. J Hum Genet 57, 515–522 (2012). https://doi.org/10.1038/jhg.2012.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.61
Keywords
This article is cited by
-
Biochemistry—Not Oncogenes—May Demystify and Defeat Cancer
Oncology and Therapy (2023)
-
Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood
Scientific Reports (2016)
