
Abstract
The β-hexosaminidase A (HEXA) mutations in the first reported cases of infantile Tay–Sachs disease in the Persian population were identified in two unrelated consanguineous families. The clinical diagnoses of the affected infants were confirmed by their markedly deficient levels of HEXA activity in plasma or peripheral leukocytes. The specific causative mutation in each family was determined by sequencing the HEXA alleles in both sets of related parents. Two mutations were identified: c.1A>G (p.MIV), which obliterated the initiating methionine in codon 1, and c.1177C>T (p.R393X), which predicted a termination codon or nonsense mutation.
Similar content being viewed by others


Introduction
Infantile Tay–Sachs disease (TSD) is a progressive neurodegenerative disorder due to the deficient activity of the lysosomal enzyme β-hexosaminidase A (HEXA), which results in the neuronal accumulation of GM2 ganglioside.1, 2 This autosomal recessive disease is characterized by onset at 4–8 months of life of neurological involvement, progressive psychomotor retardation, followed by paralysis QJ;and blindness. The cherry-red spot, due to lipid-laden ganglion cells, is the typical ophthalmologic feature. Affected children rarely survive beyond 5 years of age. In addition, juvenile- and adult-onset phenotypes have been described, which have a more protracted clinical course.3
The prevalence of TSD in general population is about 1 in 200 000 births, compared to 1 in 2500–3900 among Ashkenazi Jews.4, 5, 6 As there is no treatment for TSD, efforts have been focused on prevention by prenatal carrier identification and genetic counseling for carrier couples.5, 6 TSD screening programs, designed to identify disease carriers in Ashkenazi Jewish populations, have successfully reduced the occurrence of the disease in this population.5 We report the first cases of TSD in the Persian (Iranian) population and identify the disease-causing HEXA mutations in two unrelated families.
Case presentations
Case 1
The female proband presented at 16 months of age with weakness and motor retardation. Her parents were first cousins once removed. She was normal until 14 months when her parents noted motor problems and she lost the ability to walk by 16 months. At 24 months, she could not sit unaided, and at 26 months she had her first convulsion.
Electromyography showed mild upper motor neuron involvement, but no evidence of a peripheral neuropathy or involvement of lower motor neurons. The electroencephalography was abnormal. Ophthalmological examination revealed the macular cherry-red spot and bilateral optic nerve atrophy. Abdominal ultrasonography was normal. She did not have any hearing problems.
On the basis of these findings, the diagnosis of infantile GM2 gangliosidosis or TSD was suggested and confirmed by demonstrating markedly deficient activity of leukocyte HEXA (1.3 nmol h−1 mg−1; normal range: 90–260 nmol h−1 mg−1).
Cases 2 and 3
The probands were brothers from an Iranian family. The parents were third-degree relatives and their first child was a healthy girl. The patients were born after uneventful and full-term pregnancies. Case 2 was normal until 10 months of age when he experienced his first seizure. He steadily deteriorated and lost his vision, hearing and motor ability by 20 months. In the second year of life, myoclonic seizures became medically uncontrollable and he died at 32 months. Case 3 had some similar features but did not experience convulsions. He presented at 12 months of age with hypotonia and neurodevelopmental retardation. He lost his vision at the age of 18 months. Fundoscopy revealed the typical macular ‘cherry-red spot’. The diagnosis was confirmed by demonstration of a severe deficiency of plasma HEXA activity (0.07 mU ml−1; normal range: 0.47–2.60 mU ml−1). The patient died at 18 months of age.
Materials and methods
Molecular analysis of the HEXA gene
Genomic DNA was extracted from peripheral leukocytes by standard procedures. All HEXA exons and at least 20 bp of flanking sequence were amplified in 12 PCR mixtures using the primers in Table 1. Amplification was performed for each fragment in a 25-μl final volume containing 50 ng of genomic DNA, 0.2 mM of each dNTP, 0.2 μM of each primer, 1.0 U of Platinum Taq polymerase (Invitrogen Corp, Carlsbad, CA, USA), 1.5 mM MgCl2 and 2.5 μl of 10 × buffer for 5 min at 95 °C, followed by 35 cycles of amplification consisting of 30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C, and a final elongation of 7 min at 72 °C in an ABI 9700 Thermal Cycler (Applied Biosystems, Foster City, CA, USA). PCR products were then purified and sequenced using ABI PRISM Big Dye Terminator Cycle Sequencing (Applied Biosystems) on an ABI 3730xl automated sequencer (Applied Biosystems). Data were analyzed using ‘Sequencher’ software (Gene Codes Corporation, Ann Arbor, MI, USA).
Results
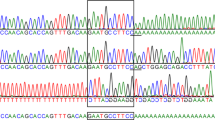
Molecular analysis of the HEXA gene in the parents from case 1 revealed that both were heterozygous for c.1177C>T (p.R393X), a nonsense mutation in codon 393. DNA from the proband was not available; homozygosity for this mutation predicts a premature termination of the enzyme protein.
In cases 2 and 3, sequencing of their genomic DNAs revealed homozygosity for a missense mutation c.1A>G, which altered the initiation methionine to a valine.
Discussion
Here, we report the first cases of TSD in the Iranian population. The HEXA mutations in three probands from two consanguineous Persian families were identified. The c.1A>G mutation was first described in an American black TSD patient7 and in a 2-year-old black child with juvenile-onset TSD disease whose other mutation Y37N (c.109T>A) had residual activity consistent with the juvenile-onset phenotype.8 Our patient is the first non-black TSD patient.
In cases 2 and 3, the HEXA mutation c.1177C>T in exon 11 caused the nonsense mutation p.R393X, which occurred at CpG dinucleotide, a known hot spot for mutations.9 This mutation was initially identified in a French infant with infantile TSD10 and was also described in a Turkish infant.11
To date, more than 130 mutations in the HEXA gene have been characterized that cause TSD.3, 12 There are ethnic specific mutations responsible for TSD in certain populations (that is, Scott et al.6), but most mutations are sporadic and frequently found in diverse populations.3 In the Ashkenazi Jewish population, three ‘founder’ mutations account for 98% of all the mutant alleles.12, 13 These include a four-base duplication c.1274_1277dupTATC (81%), the splicing mutation c.1421+1G>C (IVS12+1G>C; 15%) and a later-onset mutation c.805G>A (p.G269S; 2%).6, 12 In contrast, the major mutations in non-Jewish populations include the duplication c.1274_1277dupTATC and the missense mutation G269S that only occur at frequencies of 30 and 5%, respectively.13
As there is no effective treatment for TSD, current efforts are focused on screening populations to identify disease at-risk carrier couples,14 and then offering prenatal diagnosis. Identification of mutations in the Persian population will have direct application to this effort.
References
Okada, S. & O’Brien, J. S. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science 165, 698–700 (1969).
Sandhoff, K. Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 4, 351–354 (1969).
Gravel, R., Kabak, M. M., Proia, R. L., Sandhoff, K., Suzuki, K. & Suzuki, K. In The Metabolic and Molecular Basis of Inherited Disease (Scriver, C., Beaudet, A.L., Valle, D., Sly, W.S., Childs, B., Kinzler, K.W., Vogelstein, B. (eds)) 3827–3876 (McGraw Hill, New York, 2001).
Petersen, G. M., Rotter, J. I., Cantor, R. M., Field, L. L., Greenwald, S., Lim, J. S. et al. The Tay-Sachs disease gene in North American Jewish populations: geographic variations and origin. Am. J. Hum. Genet. 35, 1258–1269 (1983).
Kaback, M. M. Population-based genetic screening for reproductive counseling: the Tay-Sachs disease model. Eur. J. Pediatr. 159 (Suppl 3), S192–S195 (2000).
Scott, S. A., Edelmann, L., Liu, L., Luo, M., Desnick, R. J. & Kornreich, R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum. Mutat. 31, 1240–1250 (2010).
Mules, E. H., Hayflick, S., Miller, C. S., Reynolds, L. W. & Thomas, G. H. Six novel deleterious and three neutral mutations in the gene encoding the alpha-subunit of hexosaminidase A in non-Jewish individuals. Am. J. Hum. Genet. 50, 834–841 (1992).
Paciorkowski, A. R., Sathe, S., Zeng, B. J., Torres, P., Rosengren, S. S. & Kolodny, E. Juvenile-onset G(M2)-gangliosidosis in an African-American child with nystagmus. Pediatr. Neurol. 38, 284–286 (2008).
Cooper, D. N. & Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 78, 151–155 (1988).
Akli, S., Chelly, J., Lacorte, J. M., Poenaru, L. & Kahn, A. Seven novel Tay-Sachs mutations detected by chemical mismatch cleavage of PCR-amplified cDNA fragments. Genomics 11, 124–134 (1991).
Ozkara, H. A. & Navon, R. At least six different mutations in HEXA gene cause Tay-Sachs disease among the Turkish population. Mol. Genet. Metab. 65, 250–253 (1998).
Mahuran, D. J. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim. Biophys. Acta. 1455, 105–138 (1999).
Kaback, M., Lim-Steele, J., Dabholkar, D., Brown, D., Levy, N. & Zeiger, K. Tay-Sachs disease—carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970–1993. The International TSD Data Collection Network. JAMA 270, 2307–2315 (1993).
Desnick, R. J. & Kaback, M. M. Advances in Genetics: Tay-Sachs Disease (Academic Press: San Diego, CA, 2001).
Acknowledgements
We thank Ms Irina Nazarenko for her excellent technical assistance in sequencing the HEXA gene.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haghighi, A., Rezazadeh, J., Shadmehri, A. et al. Identification of two HEXA mutations causing infantile-onset Tay–Sachs disease in the Persian population. J Hum Genet 56, 682–684 (2011). https://doi.org/10.1038/jhg.2011.78
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.78
