
Abstract
Methylcrotonylglycinuria (MCG) is an inborn error of leucine catabolism and results from the deficiency of 3-methylcrotonyl-CoA carboxylase. Patients with MCG show a highly variable clinical phenotype, ranging from asymptomatic to severe. With the introduction of newborn screening using tandem mass spectrometry, most patients with MCG are identified in their asymptomatic neonatal periods. Owing to their fair clinical outcomes, there exists a controversy over the need for aggressive medical intervention or even for newborn screening for MCG. Our study, reporting 11 Korean MCG patients from nine unrelated families, who were identified by newborn screening or family member testing and normally developed without experiencing an metabolic crisis during the follow-up period of 2.6±1.96 years (range, 1–10 years), indicates that the aggressive medical intervention might not be needed at least for the MCG patients identified by screening program in asymptomatic period. A total of six MCCC2 mutations, but no MCCC1 mutation, were identified in 17 of 18 alleles (94.4%). p.D280Y was identified in the 12/18 alleles (66.7%), indicating a founder effect. Moreover, the rest five variants, p.S342K, p.Q496H, p.P552S, p.T556A and p.P459S, were all previously unreported. The results of our study indicate that the distinct molecular genetic characteristics exist in Korean MCG patients.
Similar content being viewed by others

Main
Methylcrotonylglycinuria (MCG; OMIM 210200) is an inborn error of leucine metabolism and results from 3-methylcrotonyl-CoA carboxylase (MCCC; E. C.6.4.1.4) deficiency.1 MCCC is a mitochondrial enzyme that converts 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA, a critical step for the metabolism of leucine and isovaleric acid.2 The enzyme contains α and β subunits. The MCCC1 gene encodes α subunit harboring a covalently bound biotin, which is essential for the ATP-dependent carboxylation, whereas MCCC2 encodes β subunit that possess carboxyltransferase activity.3 In MCG, 3-methylcrotonyl-CoA accumulates because of the defective MCCC and is metabolized to hydroxyisovaleric acid (3-HIVA) and 3-methylcrotonylglycine (3-MCG), the elevations of which are the hallmark for the diagnosis of MCG. MCG is inherited as an autosomal recessive trait.1 MCG used to be considered as a rare metabolic disorder. However, after neonatal screening test using tandem mass spectrometry was introduced, it is now one of the most common metabolic disorders, with the estimated prevalence of 1 : 50 000.4, 5, 6 Currently, the exact prevalence of MCG is not known in Korea, but there is a report that 1 patient was identified out of about 79 000 newborns screened in Korea.7
Patients with MCG show a highly variable clinical phenotype, ranging from asymptomatic to severe, and they usually show normal growth and development without an experience of acute metabolic crisis, even without treatment.8, 9 However, a few reports underscore the importance of medical intervention because some patients can experience metabolic decompensation accompanying various stressful events including infection, and manifest a lot of symptoms such as frequent infections, feeding difficulty, vomiting, lethargy, apnea, hypotonia, seizure, mental retardation and death in the situation of metabolic crisis.9, 10, 11, 12, 13 Therefore, there still exists a controversy over the necessity of aggressive medical intervention for MCG, or even the need for neonatal screening for MCG. Currently, newborn screening tests using tandem mass spectrometry are performed in more than 90% of the Korean newborns annually. This study was performed to report the clinical manifestations of Korean MCG patients and their clinical courses, which can help to build up the appropriate strategies for monitoring MCG patients. In addition, by revealing their mutation spectrums, our study indicates that the distinct molecular genetic characteristics exist in Korean patients with MCG.
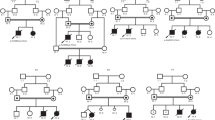
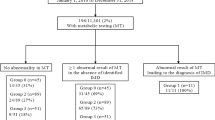
Between January 2005 and December 2010, 11 patients from nine unrelated Korean families were diagnosed as MCG at the Asan Medical Center, Seoul, Korea (Table 1). Nine patients were identified by neonatal screening program performed within 1 week of birth, whereas the rest two patients by family member screening. The mean values of urine 3-HIVA and 3-MCG were 496.8±506.9 mmol per mmol Cr (nl., 0.7–14.4 mmol per mmol Cr) and 459.6±554.2 mmol per mmol Cr (nl., 0.0 mmol per mmol Cr), respectively, which were elevated in all patients.
Total genomic DNA was isolated from peripheral white blood cells, and all the 19 exons of MCCC1 and 17 exons of MCCC2 and their respective exon–intron boundaries were analyzed in each patient. We could not find a mutation in the MCCC1 gene but only in the MCCC2 gene. A total of six MCCC2 mutations were identified in the 17 out of 18 alleles (94.4%) from the nine unrelated Korean families, which were all missense mutations (Table 1). Of note, c.838G>T (p.D280Y), a previously reported mutation in a Japanese patient with MCG by Uematsu et al,14 was the most common mutation in our patients. p.D280Y was identified in the 12/18 alleles (66.7%), indicating a founder effect. In the report by Uematsu et al.,14 this variant was only found in 1/6 alleles (16.7%). Currently, it is unknown whether p.D280Y is the common mutation in other East-Asian countries including Japan and China, as in Korea. Moreover, the rest five variants, c.1025G>A (p.S342K), c.1375C>T (p.P459S), c.1448G>C (p.Q496H), c.1654C>T (p.P552S) and c.1666A>G (p.T556A), were previously unreported. All these variants except p.Q496H were predicted as mutations by in silico analysis, but p.Q496 is evolutionally conserved as well. All the five variants were not identified in 50 normal Korean controls tested. Although their functional effects were not investigated, these variants are expected to cause MCG as a member of compound heterozygotes. All these results suggest that the distinct molecular genetic characteristics exist in Korean patients with MCG. Further investigation might be needed for the identification of molecular genetic characteristics of MCG in other East-Asian populations, which will help to understand the ethnic diversities.
Considering the accumulating metabolies including 3-HIVA and 3-MCG can cause the hyperammonemia because of suppression of urea cycle and cause hypoglycemia, metabolic acidosis, free carnitine depletion and ketoacidosis because of suppression of fatty acid metabolism or amino-acid metabolism,12, 15 we commenced the treatments including L-carnitine supplement with leucine-restricted milk formula to all patients at diagnosis. However, five patients were decided to discontinue therapy because of poor adherence. During the follow-up period of 2.6±1.96 years (range, 1–10 years), all the patients normally developed and did not experience any event of metabolic crash even during febrile illness. Their growth profiles were all normal at the latest evaluation, 4.8±2.92 years of age (range, 0.9–9.8 years of age); the median standard deviation score of body weight and height was 0.53±0.55 (range, −1.05 to 1.89) and 0.54±0.83 (−1.09 to 2.12), respectively (Table 1). However, there exist some limitations of our study, because the number of patients is small and the follow-up periods are relatively short. The fair clinical outcomes of our patients might be related to the fact that all of our patients have been identified by screening tests in asymptomatic period and a half of our patients are still on L-carnitine supplementation with dietetic therapy.
In conclusion, although a controversy still exists, our report indicates that aggressive medical intervention might not be needed at least for the patients with MCG, who were identified by screening program in asymptomatic period. As the first study reporting the mutation spectrum of Korean patients with MCG, our study reveals the distinct molecular genetic characteristics in Korean MCG patients.
References
Gallardo, M. E., Desviat, L. R., Rodriguez, J. M., Esparza-Gordillo, J., Perez-Cerda, C., Perez, B. et al. The molecular basis of 3-methylcrotonylglycinuria, a disorder of leucine catabolism. Am. J. Hum. Genet. 68, 334–46 (2001).
Moss, J. & Lane, M. D. The biotin-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 35, 321–442 (1971).
Chu, C. H. & Cheng, D. Expression, purification, characterization of human 3-methylcrotonyl-CoA carboxylase (MCCC). Protein Expr. Purif. 53, 421–7 (2007).
Koeberl, D. D., Millington, D. S., Smith, W. E., Weavil, S. D., Muenzer, J., McCandless, S. E. et al. Evaluation of 3-methylcrotonyl-CoA carboxylase deficiency detected by tandem mass spectrometry newborn screening. J. Inherit. Metab. Dis. 26, 25–35 (2003).
Liu, W., Icitovic, N., Shaffer, M. L. & Chase, G. A. The impact of population heterogeneity on risk estimation in genetic counseling. BMC Med. Genet. 5, 18 (2004).
Stadler, S. C., Polanetz, R., Maier, E. M., Heidenreich, S. C., Niederer, B., Mayerhofer, P. U. et al. Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: population heterogeneity of MCCA and MCCB mutations and impact on risk assessment. Hum. Mutat. 27, 748–59 (2006).
Yoon, H. R., Lee, K. R., Kang, S., Lee, D. H., Yoo, H. W., Min, W. K. et al. Screening of newborns and high-risk group of children for inborn metabolic disorders using tandem mass spectrometry in South Korea: a three-year report. Clin. Chim. Acta 354, 167–80 (2005).
Dantas, M. F., Suormala, T., Randolph, A., Coelho, D., Fowler, B., Valle, D. et al. 3-Methylcrotonyl-CoA carboxylase deficiency: mutation analysis in 28 probands, 9 symptomatic and 19 detected by newborn screening. Hum. Mutat. 26, 164 (2005).
Bannwart, C., Wermuth, B., Baumgartner, R., Suormala, T. & Weismann, U. N. Isolated biotin-resistant deficiency of 3-methylcrotonyl-CoA carboxylase presenting as a clinically severe form in a newborn with fatal outcome. J. Inherit. Metab. Dis. 15, 863–8 (1992).
Baumgartner, M. R., Almashanu, S., Suormala, T., Obie, C., Cole, R. N., Packman, S. et al. The molecular basis of human 3-methylcrotonyl-CoA carboxylase deficiency. J. Clin. Invest. 107, 495–504 (2001).
Baykal, T., Gokcay, G. H., Ince, Z., Dantas, M. F., Fowler, B., Baumgartner, M. R. et al. Consanguineous 3-methylcrotonyl-CoA carboxylase deficiency: early-onset necrotizing encephalopathy with lethal outcome. J. Inherit. Metab. Dis. 28, 229–33 (2005).
Dirik, E., Yis, U., Pasaoglu, G., Chambaz, C. & Baumgartner, M. R. Recurrent attacks of status epilepticus as predominant symptom in 3-methylcrotonyl-CoA carboxylase deficiency. Brain Dev. 30, 218–20 (2008).
Ficicioglu, C. & Payan, I. 3-Methylcrotonyl-CoA carboxylase deficiency: metabolic decompensation in a noncompliant child detected through newborn screening. Pediatrics 118, 2555–6 (2006).
Uematsu, M., Sakamoto, O., Sugawara, N., Kumagai, N., Morimoto, T., Yamaguchi, S. et al. Novel mutations in five Japanese patients with 3-methylcrotonyl-CoA carboxylase deficiency. J. Hum. Genet. 52, 1040–1043 (2007).
Roschinger, W., Millington, D. S., Gage, D. A., Huang, Z. H., Iwamoto, T., Yano, S. et al. 3-Hydroxyisovalerylcarnitine in patients with deficiency of 3-methylcrotonyl CoA carboxylase. Clin. Chim. Acta 240, 35–51 (1995).
Acknowledgements
We thank our patients and their family members for their participation in this study. This study was supported in part by Grant A080588 from the Korean Ministry of Health, Welfare and Family Affairs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Jung, CW., Lee, B., Kim, J. et al. Uneventful clinical courses of Korean patients with methylcrotonylglycinuria and their common mutations. J Hum Genet 57, 62–64 (2012). https://doi.org/10.1038/jhg.2011.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.116
Keywords
This article is cited by
-
Primary and maternal 3‐methylcrotonyl‐CoA carboxylase deficiency: insights from the Israel newborn screening program
Journal of Inherited Metabolic Disease (2016)
-
Metabolic heritability at birth: implications for chronic disease research
Human Genetics (2014)
-
Structure and function of biotin-dependent carboxylases
Cellular and Molecular Life Sciences (2013)
