
Abstract
Autosomal dominant retinitis pigmentosa (adRP) is a common form of RP worldwide. Although rhodopsin (RHO) is the most frequently reported adRP gene in many populations, it has not been detected in patients from the Bai nationality, one of the minority ethnic groups of southwest China. In this study, we used linkage analysis and mutation screening to identify the RHO gene in a Chinese Bai family with adRP. We found that in all affected members of the Bai family, the maximum two-point logarithm of odds score obtained was 3.61 and 4.52 at a recombination fraction (θ) of zero, with markers D3S3606 and D3S1292, respectively. Haplotype analysis showed cosegregation at the 1-c region harboring the RHO gene between the two markers with the disease. Direct sequencing of RHO revealed a c.1040C>T (p.Pro347leu) mutation in exon 5, which was supported by the reaction of the restriction enzyme. Two nonpathogenic single-nucleotide polymorphisms, rs7984 and rs2269736, were found in exon 1. To the best of our knowledge, this is the first genetic analysis of a Chinese Bai family with adRP, and a known missense RHO mutation (p.Pro347leu) is responsible for it.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP) is a genetically heterogeneous disorder primarily affecting rod photoreceptors, and is characterized by progressive degeneration of the peripheral retina, night blindness, constriction of the visual field and eventual loss of visual acuity. It is the most frequent form of inherited retinopathy, with an approximate incidence of 1 in 1000 elderly Chinese people in Northern China1 and in 1 in 4000 individuals worldwide.2 RP can be transmitted as an autosomal dominant, autosomal recessive, X-linked, mitochondrial or digenic form. So far, at least 44 gene loci have been identified as being responsible for these inheritance patterns of RP (RetNet: last updated 1 October 2009).
As Dryja et al.3 first described C-to-T transition in codon 347 of rhodopsin (RHO) in patients with RP in 1990, RHO has been found to be the most common candidate gene for autosomal dominant RP (adRP).4 From the latest summary reported in RetNet page, 66 mutational sites of RHO (Figure 1), including more than 120 mutations, have been described in RP.5, 6 Variation in distribution and frequency has been observed for different mutations in RHO. Pro23His was identified as the most frequent mutation affecting North America,7 whereas it was not reported to be present in Europe and Japan.7, 8 Conversely, Pro347Leu is the mutational hot spot all over the world,9 whereas the Val345Met mutation has been reported in some populations with very low frequencies.10, 11, 12, 13 Although mutation profiles and frequencies of RHO have been reported to be variable in different populations, few studies on the genetics of RP have been reported in the Bai (Pai) nationality, one of the minority ethnic groups of China. In this study, to analyze the potential association of RHO with RP in the Chinese Bai population, and to discover more RP-causative mutations that are unlike those of other ethnic groups, we performed a linkage analysis with short tandem repeat genetic markers and mutation screening in a large Chinese Bai family with multiple members affected by adRP.

Secondary structure model of RHO. RHO consists of the apoprotein opsin −348 amino-acid residues. The protein belongs to the G-protein-coupled receptor superfamily, and the members of this family share a basic seven-transmembrane helical design. Transmembrane helices are shown as boxed sections and the location of 66 mutated residues, summarized by Mendes,5 is highlighted in red. Pro347, one of the most common RHO mutations including L328, T342, Q344, V345 and A346, belongs to class I mutation. The sequence references to GenBank NP_000530.
Materials and methods
Family ascertainment
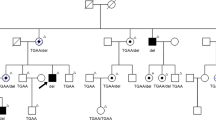
A five-generation consanguineous Bai family with RP, comprising 13 affected and 9 unaffected individuals, was identified and clinically followed up at the Department of Ophthalmology of the Guangdong General Hospital, Guangzhou, China (Figure 2). The family is from the Yunnan province of southwest China. A medical history was obtained by interviewing the family members, and routine ophthalmic evaluations were performed, including best-corrected visual acuity, slit-lamp biomicroscopy, direct funduscopy, fundus photography, visual field evaluation and electroretinography. The study protocol conformed to the tenets of the Declaration of Helsinki, and informed written consent for sample collection and analysis was obtained from each member of the family.

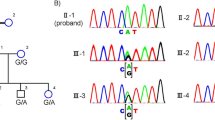
Family pedigree and haplotype diagram of the Chinese Bai family. Circles represent females and squares represent males, and a diagonal line through a symbol indicates a deceased family member. Affected members are presented as filled symbols, and unaffected members as open symbols. Black bars represent the disease allele inherited from a common ancestor. All affected individuals are heterozygous for a haploptype consisting of microsatellite markers D3S3606 and D3S1292 and the c.1040C>T mutation. The proband is labeled by an arrow.
Genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood leukocytes using an extraction kit (Purelink Genomic DNA Mini Kit, Invitrogen, Paisley, UK). To determine whether this Bai family was linked to the known adRP gene—RHO—two microsatellite markers, D3S3606 (UniSTS:726) and D3S1292 (UniSTS:73546), with an approximate by 1-cM distance on chromosome 3 were selected from the candidate region and used to genotype the family. Touchdown PCR was carried out in a 10-μl reaction mixture containing 0.5 μl genomic DNA, 1 μl 10 × PCR buffer, 0.8 μl dNTP, 0.6 μl MgSO4, 0.2 μl of each primer and 0.2 μl TAKARA Taq polymerase. Samples were incubated in My cycler PCR (BIO-Rad, Richmond, CA, USA) for 10 min at 94 °C, and for 30 s at 94 °C; the annealing temperature was programmed to start at 65 °C for 30 s and decrease by 0.5 °C every cycle; at 72 °C for 30 s, for 15 cycles; followed by 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s, for 30 cycles; a final extension at 72 °C for 10 min was carried out. PCR products were treated with Hi-Di formamide (Applied Biosystems, Foster City, CA, USA, P/N 4311320) separately and run onto capillary electrophoresis in an ABI PRISM3100 Genetic Analyzer (Applied Biosystems). The size of the allele was determined on the basis of an internal size standard (GeneScan–500 LIZ Size Standard) in each lane, and genotyping data were analyzed using the Gene Mapper version 3.5 software package (Applied Biosystems).
Two-point logarithm of odds scores were calculated at different recombination frequencies (θ=0.00, 0.05, 0.1, 0.2, 0.3, 0.4 and 0.5) using the MLINK routine of the LINKAGE software package (version 2.1.3; Cherwell Scientific Publishing Reading, Oxford, UK). The RP patient was analyzed as an autosomal dominant trait with a disease allele frequency of 0.0001. The allele frequencies for each marker were assumed to be equal, as were the recombination frequencies in males and females. Genetic penetrance was assigned at 80%. Pedigree chart and haplotypes were generated using pedigree mapping software (version 2.1.3; Cherwell Scientific Publishing, The Magdalen Centre, Oxford Science Park, Oxford, UK).
Mutation screening and reaction of restriction enzyme
Primers were designed with Primer Premier version 5.0 (Premier Biosoft International, Palo Alto, CA, USA) to amplify all coding regions and intron splice sites of RHO (GenBank NM_000539) (Table 1). Amplifications were carried out with a standard cycle in a 25-μl mixture, containing 2.5 μl 10 × PCR buffer, 2.5 μl dNTPs, 2 μl MgSO4, 0.75 μl of each primer, 0.5 μl KOD-plus-DNA polymerase (Toyobo, Osaka, Japan) and 1 μl genomic DNA, in My cycler PCR. PCR products were sequenced directly by the ABI PRISM3100 Genetic Analyzer and results were aligned with the sequence from GenBank database to perform mutation analysis.
PCR products (391 bp) of exon 5 in RHO were amplified and digested with 1 U of NlaIV (New England BioLabs, Beverly, MA, USA) restriction enzyme in a 10 μl reaction volume at 37 °C for 6 h. The resultant products were separated on an 8.0% native-PAGE gel.
Results
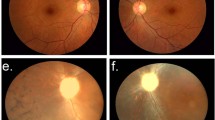
The inheritance pattern of RP in this five-generation Chinese Bai family with 22 living members is autosomal dominant (Figure 2). Thirteen affected members from this family (range 6–78 years) were phenotyped. The average age of onset was about 6–7 years. A total of 9 of 13 affected individuals had typically clinical manifestations of RP, including juvenile-onset night blindness, visual fields contraction accompanied by visual deterioration with growths, waxy pallor of the optic disc, attenuation of retinal blood vessels, bone spicule pigmentation in the midperiphery of the retina (Figure 3), and loss of the rod and cone responses in electroretinography. However, four younger affected members, individuals IV4 (13 years old), IV5 (19 years old), IV6 (9 years old) and V1 (6 years old), only complained of nyctalopia and blurred vision, and did not show any funduscopic signs of RP or changes on electroretinography. None of the unaffected individuals in this family showed typical changes in RP.

Representative fundus photographs of affected individuals. There are typical changes of adRP, including attenuated arterioles, a waxy optic and diffuse midperipheral bone spicules. (a) Proband (female, 31 years old, VOD 0.12, VOS 0.4), (b) individual III 12 (male, 45 years old, VOU 0.02). Abbreviations: VOD, visual acuity of right eye; VOS, visual acuity of left eye; VOU, visual acuity of both eyes; OD, right eye; OS, left eye.
The result of two-point linkage analysis between the disease-gene locus and the two genetic markers on chromosome 3 is shown in Table 2. At recombination frequencies of 0.000, maximum two-point logarithm of odds scores of 3.61 and 4.52 with markers D3S3606 and D3S1292, respectively, were obtained. These recombination events indicate that the disease-causing gene lies within the 1 cM interval between the two markers. Haplotype analysis for the markers in the region revealed cosegregation of alleles at markers between D3S3606 and D3S1292 with the disease in this Bai family (Figure 2).
RHO has been known to lie in the linked region and is an obvious candidate gene in this Bai family, with adRP according to the result of the linkage analysis. Therefore, mutation screening of the coding region and of the adjacent intronic regions of RHO was performed. The sequencing result showed a single base transversion in exon 5 (c.1040C>T) in the 13 affected members of the family, which was predicted to result in the protein amino-acid sequence change, p.Pro347leu. This sequence change was not observed in the nine unaffected members, who have a homozygous nucleotide at the same site (Figures 2 and 4). Restriction enzyme NlaIV digestion of RHO exon 5 PCR products from the family further supported the sequencing results and revealed the mutation to cosegregate with the disease in this Bai family (Figure 4b). In addition, two reported single-nucleotide polymorphisms (SNPs) rs7984 and rs2269736 were found to locate in the untranslated region of exon 1. The frequencies of genotype and allele of each SNP in this Bai family are shown in Table 3. Eight of 13 affected members (61.5%) were found to coexpress the homozygous genotypes AA and GG, whereas only one of nine unaffected members (11.1%) combined them.

RHO sequencing changes in affected individuals. (a) Sequence chromatograms of PCR-amplified exon 5 of RHO from the proband (IV1, top) and from a control individual (III6, bottom) from the Chinese Bai family. The arrow indicates the position of the heterozygous single base change in exon 5, c.1040C>T. (b) NlaIV digestion of exon 5 PCR products (391 bp). All affected individuals examined in the Chinese Bai family are heterozygous carriers who have all five bands at 307, 177, 130, 50 and 43 bp. The unaffected individuals are homozygous and have only the four lower bands (177, 130, 50 and 43 bp) without the upper band at 307 bp. The last two bands (50 and 43 bp) are not shown in this figure. Abbreviations: M, 20-bp DNA ladder marker (Takara); N, unaffected individualsP, affected individuals with adRP.
Discussion
Mutations associated with adRP are most commonly detected in RHO, retinal degeneration slow, inosine monophosphate dehydrogenases 1 and pre-mRNA processing factor 31 of 17 known adRP genes, which account for approximately 25, 10, 10 and 8% of adRP, respectively. We have detected retinal degeneration slow, pre-mRNA processing factor 31 and inosine monophosphate dehydrogenases 1 using direct sequencing in this Chinese Bai family with adRP. Except for a nonpathogenic four-base deletion at intron 6 of pre-mRNA processing factor 31 (IVS6-78_IVS6-75del4CACA, rs57960425), no other variations were observed in these genes (data not shown). In this study, we evaluated the linkage likelihood of RHO as a candidate gene in the Chinese Bai family with adRP using linkage analysis with microsatellite markers. Without recombination, an approximate by 1 cM region containing RHO between D3S3606 and D3S1292 was initially found to have a close linkage with the disease locus and to cosegregate with adRP. A heterozygosis single base-pair change (c.1040C>T) at exon 5 of RHO, resulting in a Pro347Leu amino-acid change in the protein, was subsequently identified by direct sequencing and confirmed by a reaction of restriction enzyme. All these results indicate that in this RP family of Bai nationality, the RHO gene is associated with adRP, and c.1040C>T (p.Pro347leu) in RHO is the causative mutation.
Of the genes known to cause adRP, mutations in RHO were most frequently detected in different ethnic groups. In Caucasian, Japanese and Chinese populations, the frequencies of RHO mutations for adRP account for approximately 25, 5.9 and 7.7%, respectively.4, 14, 15 However, it has not been reported in RP patients of Bai nationality. This minority nationality of southwest China (Yunnan province) has an ethnic background that is different from that of the Chinese Han nationality. Therefore, in this study we investigated the relationship between RHO and a Chinese Bai family with adRP, using linkage analysis for the first time. Genetic linkage analysis has a substantial role in identifying genes responsible for genetic disease. Using only the markers located in close vicinity or within the gene may improve the detection of causative genes.16 Two-point linkage analysis in our study gave evidence of close linkage between the disease-gene locus and markers D3S3606 and D3S1292. This result suggests that RHO is a major candidate gene for adRP in this Bai family because, to date, the linked region detected in this study only harbors it in known RP-causing genes.
On the basis of the result of the linkage analysis for RHO, direct sequencing as a sensitive procedure was subsequently used to reveal that there was a c.1040C>T (p.Pro347Leu) mutation in exon 5 of RHO in all affected members of the Chinese Bai family. Pro347Leu mutation is the most frequent missense change reported in six different missense mutations in Proline-347. This has been widely reported in South African, Spanish, Lithuanian, Indian, Japanese and Chinese Han populations,8, 13, 17, 18, 19 and also in the present study in a Chinese Bai population with adRP. Proline-347 has been found to be conserved in all visual pigments,3 and the Pro347Leu mutation has been shown to be involved in the pathogenic mechanism of adRP,20, 21 which indicates that it mediates some vital function of RHO. In future, therefore, the functional investigation on the role of the Pro347leu mutation in different cohorts will greatly help us understand the genetic mechanism of RP and may substantially contribute to the study on treatment.
Ando et al. revealed that no association was found between susceptibility to RP and the RHO promoter haplotype in Japanese RP patients through an assessment of their frequencies of four haplotypes constructed by five SNPs, including rs7984 and rs2269736 in the promoter region of RHO.15 In this study, two sequence alterations, c.45G>A (rs7984) and c.70A>G (rs2269736), were also found to be located in the untranslated regions of exon 1 in RHO in all members of this Bai family, and no disease-associated segregation of these changes was observed, which suggests that the changes were polymorphisms instead of disease-causing mutations. However, higher frequencies of genotypes AA of rs7984 and GG of rs2269736 were found in affected members of this family. Previous studies on thrombotic thrombocytopenic purpura have shown that, in addition to ADAMTS13 mutations, SNPs located in the ADAMTS13 coding region can influence plasmatic ADAMTS13 activity levels,22 and a combination of SNPs and a missense mutation can also modulate ADAMTS13 secretion.23 Therefore, in view of the implication of SNPs as genetic risk factors contributing to disease development and disease susceptibility, detailed investigations of a potential association between the two SNPs and adRP in the Chinese Bai population, and of the effect of combinations of the Pro347leu mutation with either or both SNPs, are needed.
The severity of RP seems to be correlated with the localization of RHO mutations. Clinically, nearly every mutation in Proline-347 has been shown to be associated with the early-onset and severe form of the disease. Similar effects have been reported for the same mutation in different ethnic populations.13 Thus, the disease feature in patients carrying the Pro347Leu mutation has been characterized by early onset, rapid progression and relative severity.24 The disease phenotypes observed in this Bai family with adRP are similar to that of other reported cohorts resulting from the Pro347Leu mutation.3, 19, 24 However, Lim et al.25 observed a co-occurrence of a novel truncating pre-mRNA processing factor 31 mutation (p.Phe262SerfsX59) and a known missense RHO mutation (p.Pro347Leu) in an affected woman of a Chinese family with adRP, which may modify the phenotype severity. Therefore, other mutations co-occurring with Pro347Leu and probably leading to the difference in clinical manifestations in the younger affected members in this Chinese Bai family will be explored in future studies.
In summary, to the best of our knowledge, this is the first genetic analysis of a Chinese Bai family with adRP. The results described in this study provide genetic data and information about the Pro347leu mutation of RHO in a Chinese Bai family, which enriches the mutation spectrum of the human RHO gene in different ethnic groups. Investigation into the genes underlying RP is potentially useful in designing genetic testing and counseling for patients. To draw definite conclusions, further studies should be performed using more sensitive methods of analysis with larger groups of RP patients of Bai nationality.
References
Xu, L., Hu, L., Ma, K., Li, J. & Jonas, J. B. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: the Beijing eye study. Eur. J. Ophthalmol. 16, 865–866 (2006).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Dryja, T. P., McGee, T. L., Hahn, L. B., Cowley, G. S., Olsson, J. E., Reichel, E. et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 323, 1302–1307 (1990).
Zhao, K., Xiong, S., Wang, L., Wang, L., Cui, Y. & Wang, Q. Novel rhodopsin mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Ophthalm. Genet. 22, 155–162 (2001).
Mendes, H. F., van, S. J., Chapple, J. P. & Cheetham, M. E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends. Mol. Med. 11, 177–185 (2005).
Cottet, S. & Schorderet, D. F. Mechanisms of apoptosis in retinitis pigmentosa. Curr. Mol. Med. 9, 375–383 (2009).
Farrar, G. J., Kenna, P., Redmond, R., McWilliam, P., Bradley, D. G., Humphries, M. M. et al. Autosomal dominant retinitis pigmentosa: absence of the rhodopsin praline—istidine substitution (codon 23) in pedigrees from Europe. Am. J. Hum. Genet. 47, 941–945 (1990).
Fujiki, K., Hotta, Y., Hayakawa, M., Sakuma, H., Shiono, T., Noro, M. et al. Point mutations of rhodopsin gene found in Japanese families with autosomal dominant retinitis pigmentosa (ADRP). Jpn. J. Hum. Genet. 37, 125–132 (1992).
Yong, R. Y., Chee, C. K. & Yap, E. P. A two-stage approach identifies a Q344X mutation in the rhodopsin gene of a Chinese Singaporean family with autosomal dominant retinitis pigmentosa. Ann. Acad. Med. Singapore 34, 94–99 (2005).
Bunge, S., Wedemann, H., David, D., Terwilliger, D. J., Born, L. I., Aulehla-Scholz, C. et al. Molecular analysis and genetic mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics. 17, 230–233 (1993).
Dryja, T. P., Hahn, L. B., Cowley, G. S., McGee, T. L. & Berson, E. L. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 88, 9370–9374 (1991).
Vaithinathan, R., Berson, E. L. & Dryja, T. P. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics 21, 461–463 (1994).
Dikshit, M. & Agarwal, R. Mutation analysis of codons 345 and 347 of rhodopsin gene in Indian retinitis pigmentosa patients. J. Genet. 80, 111–116 (2001).
Dryja, T. P., McEvoy, J. A., McGee, T. L. & Berson, E. L. Novel rhodopsin mutations Gly114Val and Gln184Pro in dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 41, 3124–3127 (2000).
Ando, Y., Ohmori, M., Ohtake, H., Ohtoko, K., Toyama, S., Usami, R. et al. Mutation screening and haplotype analysis of the rhodopsin gene locus in Japanese patients with retinitis pigmentosa. Mol. Vis. 13, 1038–1044 (2007).
Kondo, H., Tahira, T., Mizota, A., Adachi-Usami, E., Oshima, K. & Hayashi, K. Diagnosis of autosomal dominant retinitis pigmentosa by linkage-based exclusion screening with multiple locus-specific microsatellite markers. Invest. Ophthalmol. Vis. Sci. 44, 1275–1281 (2003).
Roberts, L., Ramesar, R. & Greenberg, J. Low frequency of rhodopsin mutations in South African patients with autosomal dominant retinitis pigmentosa. Clin. Genet. 58, 77–78 (2000).
Kucinskas, V., Payne, A. M., Ambrasiene, D., Jurgelevicius, V., Steponaviciute, D., Arciuliene, J. V. et al. Molecular genetic study of autosomal dominant retinitis pigmentosa in Lithuanian patients. Hum. Hered. 49, 71–74 (1999).
Chan, W. M., Yeung, K. Y., Pang, C. P., Baum, L., Lau, T. C., Kwok, A. K. et al. Rhodopsin mutations in Chinese patients with retinitis pigmentosa. Br. J. Ophthalmol. 85, 1046–1048 (2001).
Chen, J., Makino, C. L., Peachey, N. S., Baylor, D. A. & Simon, M. I. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science 267, 374–377 (1995).
Mendez, A., Burns, M. E., Roca, A., Lem, J., Wu, L. W., Simon, M. I. et al. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28, 153–164 (2000).
Kokame, K., Matsumoto, M., Soejima, K., Yagi, H., Ishizashi, H., Funato, M. et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. USA 99, 11902–11907 (2002).
Plaimauer, B., Fuhrmann, J., Mohr, G., Wernhart, W., Bruno, K., Ferrari, S. et al. Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. Blood 107, 118–125 (2006).
Berson, E. L., Rosner, B., Sandberg, M. A., Weigel-DiFranco, C. & Dryja, T. P. Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am. J. Ophthalmol. 111, 614–623 (1991).
Lim, K. P., Yip, S. P., Cheung, S. C., Leung, K. W., Lam, S. T. & To, C. H. Novel PRPF31 and PRPH2 mutations and co-occurrence of PRPF31 and RHO mutations in Chinese patients with retinitis pigmentosa. Arch. Ophthalmol. 127, 784–790 (2009).
Acknowledgements
We thank the family members for their participation in this study. We are grateful to Dr Xuchao Zhang and Dr Ailin Guo from the Guangdong Academy of Medical Sciences for their excellent professional assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Guo, H., Qin, Y., Meng, Q. et al. Linkage analysis and mutation screening of the rhodopsin gene in a Chinese Bai family with autosomal dominant retinitis pigmentosa. J Hum Genet 55, 571–576 (2010). https://doi.org/10.1038/jhg.2010.68
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.68
