
Abstract
Hereditary paraganglioma (PGL) is characterized by the development of highly vascularized paraganglionic tumors as a result of germline mutations in the SDHB, SDHC or SDHD subunit genes of succinate dehydrogenase (SDH; mitochondrial complex II), or in the Von Hippel–Lindau tumor-suppressor gene. Although many PGL mutations have been described, gross SDHD deletions have not yet been implicated as founder mutations and are rarely characterized at the DNA sequence level. We investigated the genetic basis of head and neck PGLs observed in 20 subjects from two unrelated multiplex pedigrees from Austria and identified a 4944-base pair partial SDHD deletion, which escaped PCR-based detection methods. The deletion occurred between Alu elements and was present within the same haplotype context in both pedigrees, indicating a founder effect. The deletion caused tumors only after a paternal transmission similar to other conventional SDHD mutations, suggesting preservation of genomic imprinting mechanisms operating at this locus. These data describe a large SDHD deletion at the genomic sequence level and indicate that gross SDHD deletions could be a founder PGL mutation in certain populations.
Similar content being viewed by others


Introduction
Paragangliomas (PGLs) are highly vascular tumors derived from the paraganglionic system, which comprises many small tissues and organs distributed from the skull base to the pelvic floor. Paraganglionic tissues mostly associate with the parasympathetic nervous system in the head and neck and cluster around major arteries and nerves. The carotid body is the major paraganglion in the head and neck. The paraganglia in the non-head and neck regions such as the adrenal medulla and the organ of Zuckerkandl are more closely associated with the sympathetic nervous system. Abdominal PGLs often secrete catecholamines and are referred to as pheochromocytomas. Normal paraganglia have an important role in organismic homeostasis against stressors, such as hypoxia, hypoglycemia and immobilization.1
Hereditary PGLs are caused by germline heterozygous inactivating mutations in the SDHB, SDHC or SDHD subunit genes of succinate dehydrogenase (SDH; mitochondrial complex II),2, 3, 4 and can occur in the Von Hippel–Lindau disease caused by VHL mutations.5 Mutations in SDHD, but not in SDHB or SDHC, cause PGL only after a paternal inheritance, suggesting an operation of genomic imprinting (inactivation) on the SDHD locus.6 To date, clinically and histolopathologically confirmed PGLs have not been documented to develop on maternal inheritance of an SDHD mutation.7 However, the molecular basis of genomic imprinting on SDHD remains unknown. Most familial head/neck PGLs (HNPs) and more than 11% of isolated HNPs carry germline mutations in one of the three SDH genes.1, 8, 9, 10 In addition, ∼8% of non-syndromic pheochromocytomas carry germline mutations in SDHB or SDHD genes.1, 11, 12 Germline mutations in SDHD seem to be more prevalent among HNPs,8, 13 whereas SDHB mutations are more common in extra-adrenal and malignant pheochromocytomas.12, 13 SDHC mutations are uncommon and are primarily linked to HNPs.10
Multiple PGL founder mutations were thus far described only in the Netherlands, where three missense SDHD mutations were responsible for nearly all familial HNPs and for at least one-fourth of the isolated cases.14 The presence of multiple founder mutations in the Netherlands has been attributed in part to the very low altitudes in this country, which could reduce the penetrance and expressivity and relax the natural selection on SDHD mutations.15 In addition, single founder SDHD mutations were identified by haplotype analyses in the United States,8 Italy,16 and were also suggested by the identification of rare SDHD mutations in unrelated cases from Spain17 and China.18 All SDHD founder mutations and most SDH mutations described thus far are single-nucleotide changes or deletions/insertions of a few base pairs identified by PCR-based techniques.19 Large germline founder deletions in SDHB and SDHC were characterized at the sequence level in multiple unrelated subjects from Spain and the United States, respectively.9, 12, 20
Complete gene deletions in SDHD were described in two studies using multiplex PCR amplification and quantification of gene exons.21, 22 These studies, however, did not characterize the precise extent of SDHD deletions by identifying the DNA sequences spanning the deletion break points. Identification of SDHD deletion break points enables quick population screening of founder mutations by conventional PCR, helps inferring deletion mechanisms and might provide new insights for regulation of the imprinted transmission of SDHD mutations. In this report, we describe identification of the first SDHD founder deletion in two extended PGL families from Austria and capture and characterize the deletion break points at the DNA sequence level.
Materials and methods
Clinical presentation
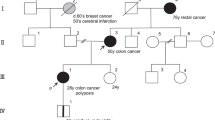
Two multi-generational PGL families (namely PGL38 and PGL101) were ascertained in Innsbruck, Austria. PGL38 and PGL101 comprised 17 and 3 affected subjects, respectively (Figure 1). DNA samples obtained from 11 affected subjects from PGL38 and 3 affected subjects from PGL101 were available for genetic study. All affected individuals had single or multiple HNPs, as diagnosed by clinical and histopathological methods. Clinical data that were available for 19 affected subjects showed development of bilateral HNPs in 15 (79%). There was no instance of maternal transmission of the mutation among the affected subjects, suggesting that the underlying mutation was located in the SDHD gene.

Pedigrees of two extended paraganglioma (PGL) families ascertained from Austria. The disease is transmitted exclusively from an affected father in all informative nuclear families. Small circles below subjects denote head/neck paraganglioma tumors (right/left sided), and crosses denote head/neck paraganglioma tumors treated by surgery. Filled or empty circles/squares denote affected or unaffected females/males, respectively.
Gene sequencing, haplotype and deletion analyses
Informed consent for molecular studies was obtained from all participants after genetic counseling. DNA was extracted from peripheral blood samples using standard methods. PCR amplification and sequencing of SDHD, SDHB and SDHC genes from peripheral blood was performed as described previously.8 To test the cosegregation of the SDHD locus among affected individuals, 11 simple tandem-repeat polymorphisms that spanned SDHD on the long arm of chromosome 11 were genotyped.
To test for the presence of a large intragenic SDHD deletion, we first performed long-range PCR by combining forward PCR primers of each exon with the reverse primers of the downstream exons. Subsequently, we pursued separation of the disease and non-disease chromosomes 11 in somatic cell hybrids and performed sequence tagged site content analysis using four stable hybrids, each containing a maternal or paternal chromosome 11 from an affected subject from each family.
Results
PCR amplification and sequencing of SDHD, SDHB and SDHC genes from peripheral blood DNA showed no mutations. As the pedigrees predominantly show HNPs transmitted by fathers, we hypothesized the presence of an unconventional mutation in the SDHD gene. Cosegregation of the SDHD locus with the disease was supported by the demonstration of a common haplotype that was shared by all genotyped affected subjects in PGL38 (n=8) and PGL101 (n=2) (Table 1).
Long-range PCR by combining forward PCR primers of each exon with the reverse primers of the downstream exons showed no aberrant bands amplified from the affected subjects.
However, in the affected chromosomes 11 separated by somatic cell hybrids in subjects, SDHD exons 1, 2 or 3 amplified but exon 4 did not amplify, suggesting a deletion spanning exon 4, thus gaining direct evidence for a disease-causing deletion. Further sequence tagged site content analyses delineated the approximate locations of break points. The deletion break-point junction was captured within a 494-bp PCR amplicon (Figure 2a), the oligonucleotide primers of which are located 5454 bp away in the normal genomic DNA sequence. The presence and zygosity of the deletion break-point junction was confirmed by a duplex PCR (Figure 2b). The deletion was present in all affected subjects, but not in 100 control chromosomes.

(a) The deletion in exon 4 spans two similar Alu repeat elements, which might have triggered an illegitimate recombination event. The sequence chromatogram of the junctional amplicon at the bottom shows the deletion junction and the neighboring Alu sequences (boxes). (b) The SDHD deletion break point and wild-type fragment are captured in a duplex PCR amplification. The PCR reaction contained a common forward oligonucleotide primer (FFD: 5′-ATGCTGTCTCACTTACTATACTCCCAAC-3′) flanking the deletion, a reverse primer (RD: 5′-ATGTCTCCATTTGCCACTCCAC-3′) from the deleted segment and a reverse primer (RFD: 5′-GGCTTAGTAGATACTCTTAGGGGCAAC-3′) flanking the deletion. The FFD and RD amplify a 787-bp wild-type amplicon, whereas FFD and RRD, located 5454 bp apart in the genomic sequence, amplify a 494-bp amplicon in deletion carriers. A, affected; C, control.
Sequencing of the break-point junction confirmed that the deletion removed exon 4, the terminal coding exon and conjoined two Alu sequences in the third intron and the 3′-flanking region, respectively. The proximal deletion break point occurred within a positively oriented partial AluJo fragment, and the distal break point occurred within a negatively oriented complete AluSq repeat. A positively oriented complete AluSg repeat, located 38 bp downstream of the proximal break point had 86% sequence identity with the AluSq repeat of the distal break point, raising the possibility that an illegitimate recombination between the AluSg and AluSq repeats precipitated the genomic deletion.
Discussion
In this study, we characterized a large exon-spanning SDHD deletion at the DNA sequence level and showed that this deletion is a founder mutation of two extended PGL families from Austria. The identification of a deletion as a founder mutation indicates that the contribution of SDHD germline mutations to PGLs is even higher than that suggested by PCR-based mutation screening techniques. Accordingly, the founder deletion is found in two of four (50%) extended Austrian PGL families investigated in this study and in an earlier work.23 The current results will enable rapid PCR screening of the founder deletion in familial, early-onset or multi-focal PGLs that show no coding mutations in PGL genes in Austria and Central Europe. Our findings support recent evidence for the necessity to perform a copy-number analysis of SDH exons in patients without point mutations or small deletions/insertions, which is facilitated by the availability of the multiplex ligation-dependent probe amplification technique for these genes. Larger germline deletions accounted for 6 and 3% of kindred with SDHB and SDHD mutations, respectively, in one large study of 358 patients with mutations.12
Notably, the deletion bears certain similarities with the previously described SDHC deletion in that it removes the terminal coding exon, spans two highly similar Alu repeat elements and occurs within a region of high Alu content.9 Although only 10% of the human genome is composed of Alu repeats, they constitute 24% of the SDHD genomic sequence between the gene transcription start site and the end of terminal coding exon, and 42% of the 5-kb long genomic sequence immediately flanking the terminal exon of SDHD, where the distal break point of the deletion occurred. As Alu-mediated recombination events are well recognized in the etiology of large germline deletions,24 high Alu content, especially at the 3′-flanking region, of SDHD might be implicated in the pathogenesis of the deletion.
In this study, the exclusive paternal transmission of the PGL disease phenotype supports a model in which the maternal SDHD copy is suppressed in the paraganglionic tissues by genomic imprinting. Congruent with this model, the SDH complex also seems to be downregulated by other mechanisms such as balancing missense polymorphisms at highly conserved amino acids in SDHA25 and the occurrence of a targeted somatic stop-codon mutation in SDHB in normal peripheral blood mononuclear cells.26 Characterization of large gene deletions at 11q23 in the vicinity of the SDHD gene might facilitate identifying genomic regions important for the imprinted transmission of the SDHD gene. As previously shown in the imprinted Prader-Willi/Angelman loci,27 germline deletions of the imprint switch regions could prevent acquisition of maternal- or paternal-specific imprinted patterns during sex-specific gametogenesis and could cause disease by silencing of imprinted genes or alter imprinted inheritance patterns. As the PGL phenotype is transmitted exclusively from fathers in current pedigrees, deletion rules out the presence of an imprint switch region in the deleted genomic segment.
In summary, our findings describe the first large SDHD deletion characterized at the sequence level and demonstrate that gross SDHD deletions could be founder PGL mutations in certain populations.
References
Baysal, B. E. Clinical and molecular progress in hereditary paraganglioma. J. Med. Genet. 45, 689–694 (2008).
Baysal, B. E., Ferrell, R. E., Willett-Brozick, J. E., Lawrence, E. C., Myssiorek, D., Bosch, A. et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000).
Niemann, S. & Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 26, 268–270 (2000).
Astuti, D., Latif, F., Dallol, A., Dahia, P. L., Douglas, F., George, E. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 49–54 (2001).
Gaal, J., van Nederveen, F. H., Erlic, Z., Korpershoek, E., Oldenburg, R., Boedeker, C. C. et al. Parasympathetic paragangliomas are part of the Von Hippel-Lindau syndrome. J. Clin. Endocrinol. Metab. 94, 4367–4371 (2009).
van der Mey, A. G., Maaswinkel-Mooy, P. D., Cornelisse, C. J., Schmidt, P. H. & van de Kamp, J. J. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet 2, 1291–1294 (1989).
Neumann, H. P. & Erlic, Z. Maternal transmission of symptomatic disease with SDHD mutation: fact or fiction? J. Clin. Endocrinol. Metab. 93, 1573–1575 (2008).
Baysal, B. E., Willett-Brozick, J. E., Lawrence, E. C., Drovdlic, C. M., Savul, S. A., McLeod, D. R. et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J. Med. Genet. 39, 178–183 (2002).
Baysal, B. E., Willett-Brozick, J. E., Filho, P. A., Lawrence, E. C., Myers, E. N. & Ferrell, R. E. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J. Med. Genet. 41, 703–709 (2004).
Schiavi, F., Boedeker, C. C., Bausch, B., Peczkowska, M., Gomez, C. F., Strassburg, T. et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 294, 2057–2063 (2005).
Neumann, H. P., Bausch, B., McWhinney, S. R., Bender, B. U., Gimm, O., Franke, G. et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 346, 1459–1466 (2002).
Ricketts, C. J., Forman, J. R., Rattenbury, E., Bradshaw, N., Lalloo, F., Izatt, L. et al. Tumour risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 31, 41–51 (2009).
Neumann, H. P., Pawlu, C., Peczkowska, M., Bausch, B., McWhinney, S. R., Muresan, M. et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951 (2004).
Taschner, P. E., Jansen, J. C., Baysal, B. E., Bosch, A., Rosenberg, E. H., Brocker-Vriends, A. H. et al. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer 31, 274–281 (2001).
Astrom, K., Cohen, J. E., Willett-Brozick, J. E., Aston, C. E. & Baysal, B. E. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum. Genet. 113, 228–237 (2003).
Simi, L., Sestini, R., Ferruzzi, P., Gagliano, M. S., Gensini, F., Mascalchi, M. et al. Phenotype variability of neural crest derived tumours in six Italian families segregating the same founder SDHD mutation Q109X. J. Med. Genet. 42, e52 (2005).
Velasco, A., Palomar-Asenjo, V., Ganan, L., Catasus, L., Llecha, N., Panizo, A. et al. Mutation analysis of the SDHD gene in four kindreds with familial paraganglioma: description of one novel germline mutation. Diagn. Mol. Pathol. 14, 109–114 (2005).
Lee, S. C., Chionh, S. B., Chong, S. M. & Taschner, P. E. Hereditary paraganglioma due to the SDHD M1I mutation in a second Chinese family: a founder effect? Laryngoscope 113, 1055–1058 (2003).
Bayley, J. P., Devilee, P. & Taschner, P. E. The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med. Genet. 6, 39 (2005).
Cascon, A., Landa, I., Lopez-Jimenez, E., Diez-Hernandez, A., Buchta, M., Montero-Conde, C. et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J. Med. Genet. 45, 233–238 (2008).
McWhinney, S. R., Pilarski, R. T., Forrester, S. R., Schneider, M. C., Sarquis, M. M., Dias, E. P. et al. Large germline deletions of mitochondrial complex II subunits SDHB and SDHD in hereditary paraganglioma. J. Clin. Endocrinol. Metab. 89, 5694–5699 (2004).
Mannelli, M., Castellano, M., Schiavi, F., Filetti, S., Giacche, M., Mori, L. et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 94, 1541–1547 (2009).
Fish, J. H., Klein-Weigel, P., Biebl, M., Janecke, A., Tauscher, T. & Fraedrich, G. Systematic screening and treatment evaluation of hereditary neck paragangliomas. Head Neck 29, 864–873 (2007).
Stankiewicz, P. & Lupski, J. R. The genomic basis of disease, mechanisms and assays for genomic disorders. Genome Dyn. 1, 1–16 (2006).
Baysal, B. E., Lawrence, E. C. & Ferrell, R. E. Sequence variation in human succinate dehydrogenase genes: evidence for long-term balancing selection on SDHA. BMC Biol. 5, 12 (2007).
Baysal, B. E. A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia. PLoS One 2, e436 (2007).
Buiting, K., Saitoh, S., Gross, S., Dittrich, B., Schwartz, S., Nicholls, R. D. et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 9, 395–400 (1995).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Janecke, A., Willett-Brozick, J., Karas, C. et al. Identification of a 4.9-kilo base-pair Alu-mediated founder SDHD deletion in two extended paraganglioma families from Austria. J Hum Genet 55, 182–185 (2010). https://doi.org/10.1038/jhg.2009.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.142
