
Abstract
We report a 67-year-old Japanese woman with ataxia with oculomotor apraxia type 2 (AOA2). She was born to consanguineous parents and showed a teenage onset, a slowly progressive cerebellar ataxia and sensory-motor neuropathy and an elevated level of serum α-fetoprotein (AFP). All of these clinical features were consistent with typical AOA2. She lacked oculomotor apraxia, as frequently observed in previously reported AOA2 patients. She was homozygous for a novel nonsense mutation, Glu385Ter (E385X), in the senataxin gene (SETX). To our knowledge, this is the fifth Japanese family with genetically confirmed AOA2. The mutations in SETX in Japanese AOA2 families are heterogeneous, except for M274I, which has been found in two unrelated families. More extensive screening by serum AFP followed by molecular genetic analysis of SETX in patients with Friedreich’s ataxia-like phenotype may show that AOA2 is more common in Japan than previously thought.
Similar content being viewed by others

Main
Ataxia with oculomotor apraxia type 2 (AOA2) is a rare form of autosomal-recessive cerebellar ataxias associated with axonal sensory-motor neuropathy.1 The disease-causing gene (SETX) encodes a protein, senataxin, with a homology to the fungal Sen1p protein.2 SETX, which is composed of 2677 amino acids, contains a DNA/RNA helicase domain (1931–2446 amino acids) near the C terminus, and is likely to have important roles in DNA repair and transcription, and RNA processing.2, 3, 4 The N-terminal domain (64–593 amino acids) of SETX is considered a putative protein interaction domain essential for proper localization of SETX.5
To date, more than 50 mutations have been identified in AOA2 families worldwide.1, 2, 6, 7, 8, 9, 10, 11, 12 The mutations are distributed across the entire gene, without any specific mutation hot spots. Most of the mutations are frameshift, or nonsense mutations, supporting that a loss of function of SETX is critical for the development of AOA2.
In this study, we describe a 67-year-old Japanese patient with AOA2 who carried a novel homozygous nonsense mutation, Glu385Ter (E385X), in SETX. Consistent with a prolonged clinical course, she showed prominent fiber type grouping in muscle biopsy. This study identifies the fifth Japanese family with genetically confirmed AOA22, 8, 13, 14, 15 (H Takashima, personal communication).
Case report
The patient was a 67-year-old Japanese woman, who was born to consanguineous parents and attained normal developmental milestones during infancy. At ∼17 years old, she began falling frequently while walking. At age 20, she was referred to the hospital because of an unsteady gait. Soon thereafter, she was diagnosed with cerebellar ataxia, and by age 22, she could not walk without assistance. At ∼40 years old, she began to show speech disturbance and became clumsy with her hands.
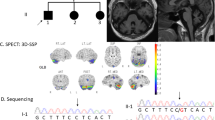
She had her first extensive medical examinations at age 46. Neurological examination showed horizontal gaze nystagmus and dysarthria, but not oculomotor apraxia. In addition, she showed distal dominant muscle atrophy and weakness, hypesthesia of the glove and stocking type, loss of position and vibration sense, and limb and truncal ataxia. Deep tendon reflexes were decreased in her upper extremities, and absent in her lower extremities. Pyramidal tract signs were not evident. She was not able to stay standing even when holding a handrail. Routine blood and cerebrospinal fluid examinations were normal. Magnetic resonance imaging of the brain showed atrophy in the cerebellar hemispheres and vermis, but not in the brainstem. Needle electromyogram showed high amplitude and long-duration motor potentials in the muscles examined. Ulnar nerve motor conduction velocity was 48 m s−1, and compound-muscle action potentials of the lower limbs could not be obtained. Sensory nerve conduction velocities were reduced in both the upper and lower limbs. A quadriceps muscle biopsy showed large fiber type grouping (Figure 1a), and a sural nerve biopsy showed a severe loss of myelinated fibers without onion bulb formation. At this time, she was diagnosed as having a combined phenotype of spinocerebellar ataxia and sensory-motor neuropathy.

The muscle biopsy from the quadriceps muscles shows a large fiber type grouping (a, routine ATPase stain, bar=100 μm, age 46 years). Distal muscle wasting in the hands and fixed flexion of the PIP and DIP joints are seen (b, age 67 years). Clubfoot and drop foot deformities are observed (c, age 67 years). Magnetic resonance images show atrophy in the cerebral hemispheres and vermis (d, age 67 years) and the sequencing results of SETX indicate a homozygous E385X mutation (e). The asterisk indicates the position of a G-to-T substitution. For the cDNA, the nucleotide number +1 corresponds to the A of the ATG translation initiation codon.
At age 67, her condition was re-evaluated in our hospital. Her cognitive function was well preserved. She showed horizontal gaze nystagmus and disturbance of smooth pursuits. Her speech was slurred, but dysphagia was absent. Head tremor was observed. Distal muscle wasting and weakness, with finger and foot deformities, were evident (Figures 1b and c). Routine blood studies were normal, but serum α-fetoprotein (AFP) was markedly high (123.0 ng ml−1, normal range 0–10 ng ml−1). Magnetic resonance imaging of the brain showed a mega-cisterna magna and atrophy of the cerebellar hemispheres and vermis (Figure 1d), while that of the spinal cord and abdominal ultrasonography showed no abnormal findings.
As the clinical and laboratory features in this patient were consistent with AOA2, we searched for a mutation in SETX in this patient according to the previous report.8 The results showed that the patient carried a novel homozygous nonsense mutation, E385X (c. 1153G → T in exon 8) (Figure 1e) in SETX, whereas her two unaffected siblings were heterozygous for this mutation.
Discussion
AOA2 is an early-onset cerebellar ataxia of autosomal-recessive inheritance caused by mutations in SETX.1, 2 It generally starts with cerebellar ataxia and then axonal sensory-motor neuropathy overlays with disease progression. Oculomotor apraxia is included in the disease acronym, but it is not a consistent feature of the disease.1, 2 Less-common clinical features include pyramidal signs and movement disorders such as dystonia, choreic movements and tremor. Intelligence, as well as bulbar and respiratory functions, is relatively unaffected in AOA2 patients. Almost all patients with AOA2 (>90%) show elevated serum AFP, which serves as a good diagnostic marker for AOA2.1, 2 Despite a large heterogeneity in SETX mutations in more than 50 AOA2 families with various ethnic backgrounds, its clinical findings and course are relatively consistent.
The patient described in this study became wheelchair-bound in ∼5 years after onset. This is a shorter period than previously reported.6, 7, 9, 12 However, despite rapid deterioration of functional independence after onset, her overall clinical course was very protracted, and the patient survived into her late 60s. The prominent fiber type grouping observed in the quadriceps muscles was consistent with chronic denervation and reinnervation in peripheral nerves.
AOA2 was described initially in a Japanese family,13 and to our knowledge, the present family is the fifth with genetically proven AOA2 in Japan2, 8, 13, 14, 15 (H Takashima, personal communication). Mutations in SETX identified in Japanese AOA2 families are heterogeneous, except M271I, which has been found in two unrelated Japanese families2, 8, 15 (H Takashima, personal communication). E385X reported in this study causes an early truncation in the N-terminal domain, which is very likely to lack both N-terminal and helicase domain functions.
To date, more than 50 mutations have been identified in AOA2 and 4 mutations in its allelic disorder, amyotrophic lateral sclerosis 4 (ALS4), worldwide.1, 2, 6, 7, 8, 9, 10, 11, 12, 16 The pathogenic mechanism of SETX mutations for causing two different allelic diseases, AOA2 and ALS4, needs to be further clarified. All the four SETX mutations reported in ALS4 patients are missense mutations, three of which cluster within either DNA/RNA helicase domain or N-terminal domain.16 On the other hand, most of the AOA2 mutations are nonsense or frameshift, most likely causing a loss of function of SETX.
In Japan, autosomal-recessive cerebellar ataxia accounts for ∼1.8% of spinocerebellar ataxia patients,17 but the disease frequency of each subtype in autosomal-recessive cerebellar ataxia is unclear. A genetically confirmed Friedreich's ataxia patient has not been found in the Japanese population, and the majority of patients with Friedreich's ataxia-like phenotype may be confirmed as AOA1, AOA2, ataxia with vitamin E deficiency or autosomal-recessive spastic ataxia of Charlevoix-Saguenay if examined by genetic testing. As elevation of serum AFP concentration is highly specific for AOA2, biochemical screening followed by molecular genetic testing of SETX in patients with the FDRA-like phenotype will facilitate our understanding of the disease frequency of the subtypes in autosomal-recessive cerebellar ataxia in Japan.
References
Moreira, M.- C. & Koenig, M. Ataxia with oculomotor apraxia type 2 in GeneReviews at GeneTests: Medical Genetics Information Resource [database online] Copyright, (University of Washington, Seattle, 1993–2008). Available at http://www.genetests.org; updated 5 March 2007.
Moreira, M.- C., Klur, S., Watanabe, M., Németh, A. H., Le Ber, I., Moniz, J.- C. et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 36, 225–227 (2004).
Suraweera, A., Becherel, O. J., Chen, P., Rundle, N., Woods, R., Nakamura, J. et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 177, 969–979 (2007).
Suraweera, A., Lim, Y., Woods, R., Birrell, G. W., Nasim, T., Becherel, O. J. et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 18, 3384–3396 (2009).
Chen, Y. Z., Hashemi, S. H., Anderson, S. K., Huang, Y., Moreira, M.- C., Lynch, D. R. et al. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol. Dis. 23, 97–108 (2006).
Le Ber, I., Bouslam, N., Rivaud-Péchoux, S., Guimarães, J., Benomar, A., Chamayou, C. et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain 127, 759–767 (2004).
Duquette, A., Roddier, K., McNabb-Baltar, J., Gosselin, I., St-Denis, A., Dicaire, M.- J. et al. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann. Neurol. 57, 408–414 (2005).
Asaka, T., Yokoji, H., Ito, J., Yamaguchi, K. & Matsushima, A. Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology 66, 1580–1581 (2006).
Criscuolo, C., Chessa, L., Di Giandomenico, S., Mancini, P., Saccà, F., Grieco, G. S. et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 66, 1207–1210 (2006).
Fogel, B. L. & Perlman, S. Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology 67, 2083–2084 (2006).
Anheim, M., Fleury, M. C., Franques, J., Moreira, M.- C., Delaunoy, J. P., Stoppa-Lyonnet, D. et al. Clinical and molecular findings of ataxia with oculomotor apraxia type 2 in 4 families. Arch. Neurol. 65, 958–962 (2008).
Tazir, M., Ali-Pacha, L., M'Zahem, A., Delaunoy, J. P., Fritsch, M., Nouioua, S. et al. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J. Neurol. Sci. 278, 77–81 (2009).
Watanabe, M., Sugai, Y., Concannon, P., Koenig, M., Schmitt, M., Sato, M. et al. Familial spinocerebellar ataxia with cerebellar atrophy, peripheral neuropathy, and elevated level of serum creatine kinase, γ-globulin, and α-fetoprotein. Ann. Neurol. 44, 265–269 (1998).
Bomont, P., Watanabe, M., Gershoni-Barush, R., Shizuka, M., Tanaka, M., Sugano, J. et al. Homozygosity mapping of spinocerebellar ataxia with cerebellar atrophy and peripheral neuropathy to 9q33-34, and with hearing impairment and optic atrophy to 6p21-23. Eur. J. Hum. Genet. 8, 986–990 (2000).
Watanabe, M., Okamoto, K. & Shoji, M. Ataxia-ocular apraxia 2. Shinkei Kenkyu No Shinpo (Adv. Neurol. Sci.) 50, 371–377 (2006) (in Japanese).
Chen, Y.- Z., Bennett, C. L., Huynh, H. M., Blair, I. P., Puls, I., Irobi, J. et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135 (2004).
Tsuji, S., Onodera, O., Goto, J. & Nishizawa, M. Sporadic ataxias in Japan—a population-based epidemiological study. Cerebellum 7, 189–197 (2008).
Acknowledgements
We thank Dr Hiroshi Takashima for kindly providing us with the information on his unpublished AOA2 family. This work was supported in part by a Grant-in-Aid for Science Research from the Ministry of Education, Science and Culture, Japan, and a grant from the Research Committee for Ataxic Diseases, the Ministry of Health, Labour, and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakamura, K., Yoshida, K., Makishita, H. et al. A novel nonsense mutation in a Japanese family with ataxia with oculomotor apraxia type 2 (AOA2). J Hum Genet 54, 746–748 (2009). https://doi.org/10.1038/jhg.2009.104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.104
