
Abstract
The Mayan homeland within Mesoamerica spans five countries: Belize, El Salvador, Guatemala, Honduras and Mexico. There are indications that the people we call the Maya migrated from the north to the highlands of Guatemala as early as 4000 B.C. Their existence was village-based and agricultural. The culture of these Preclassic Mayans owes much to the earlier Olmec civilization, which flourished in the southern portion of North America. In this study, four different Mayan groups were examined to assess their genetic variability. Ten polymorphic Alu insertion (PAI) loci were employed to ascertain the genetic affinities among these Mayan groups. North American, African, European and Asian populations were also examined as reference populations. Our results suggest that the Mayan groups examined in this study are not genetically homogeneous.
Similar content being viewed by others

Introduction
The Mayans, as a group, have exhibited over the ages an extraordinary cultural cohesion despite the extent and ecological diversity of their territory, a multitude of languages and the presence of hostile groups that in the past have included the Aztecs to the north, less sophisticated groups to the south and, more recently, the Europeans and the Spanish American civilization. The highly heterogeneous geographic landscape of their territory including deserts, rain forests and snow-covered highlands could have served as barriers limiting interaction within the region. Yet, the integrity of the Mayans as a group is evident today in the strong correlation between the Mayan family of languages and culture (Coe 1992).
Neither archaeology nor linguistics has provided much insight into the origins of the Mayans. It has been theorized that the Mayans as a people settled in the highlands of what is today western Guatemala about 6,000 years ago during the late Archaic period. The fact that the Mayans shared with other Mesoamerican groups certain characteristics like hieroglyphic writing, complex calendars, a sophisticated knowledge of astronomy and human sacrifice, which were developed by the earlier Olmecs of southern Mexico, suggests that much of the Mayan complex culture derives from the Olmec mother culture. Since then the interchange of ideas among Mesoamerican peoples has been prevalent. This by itself would tend to bring about cultural homogeneity in the area.
Widespread genetic information on Native Americans has been collected and reviewed (Salzano and Callegari-Jacques 1988; Crawford 1998; Salzano 2002). The last author evaluated the molecular variation among (i.e., G st) North, Central and South Native American populations and found that the levels of inter-population variation is higher in comparison to other worldwide groups. In addition, limited variation within populations (i.e., H t) is found in American natives as a whole when HLA polymorphisms (Black 1991; Arnaiz-Villena et al. 2000), polymorphic Alu insertions (PAIs) (Novick et al. 1998; Mateus-Pereira et al. 2005), mitochondrial DNA (Bonatto and Salzano 1997; Silva et al. 2002) and Y-chromosome data (Santos et al. 1996; Bianchi et al. 1997, 1998) are examined, supporting the idea of a limited number of entries of founders into the American continent. However, the genetic information is highly heterogeneous with respect to the populations and types of genetic systems investigated (Salzano 2002). In an effort to better understand the genetics of Mesoamerican groups, we undertook this study of the Mayans. To our knowledge, this work is the first to investigate the genetic make up of several regional Mayan groups. To accomplish this, the allelic frequencies of ten PAI loci were examined. Alu repeats are the largest family of SINEs in the human genome, comprising about 1 million copies per somatic cell and representing approximately 10% of the genome of our species (Carroll et al. 2001; Donaldson et al. 2002). There are several advantages to using PAIs in human population genetic studies. First, the insertions represent unique events and reflect identity by descent. In other words, the likelihood of an insertion being completely removed leaving no detectable Alu sequences behind or an element inserted twice independently at a specific location is low (Batzer et al. 1994; Hillis 1999; York et al. 1999). Second, the ancestral state of the character is known. The absence of the element at a specific chromosomal site represents the ancestral condition. This information provides the polarity so valuable in phylogenetic analyses (Batzer et al. 1994; Martinez et al. 2005).
The principal goal of this study was to investigate the degree of genetic variability among four geographically targeted Mayan groups. Our aim was to examine whether the Mayan cultural uniformity and linguistic commonalities discussed above are also reflected in their genetic make-up. It is possible that despite their extensive communication routes throughout the empire, geographical barriers and the spatial partitioning imposed by their city-state political system have restricted gene flow for the development of unique gene pools. For this purpose, we examined the Mayan groups from the Buctzotz, Cakchiquel, Campeche and Quiche.
Materials and methods
Subjects
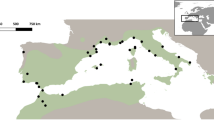
The samples were collected from autochthonous donors who could trace their specific Mayan ancestry at least two generations back to specific tribes (e.g., the Quiche). We defined individuals to each of the four Mayan populations by last name and self identification to specific tribes. These individuals were sampled in rural locations known to be inhabited exclusively by autochthonous people. These requirements minimize the possibility of collecting from admixed populations. Information concerning the geographical locations and linguistic affiliations of the groups examined in this study is given in Fig. 1 and Table 1. Six Native Americans (four Mayan and two North Americans), one European, two Asians and two Africans populations were examined. The Mayan groups have been characterized as Amerindians, while the Navajo and the Eskimo from North America are Na-Dene and Eskimo-Aleut, respectively (Greenberg 1987). These language families comprise the three major linguistic groups spoken among the natives of the New World (Greenberg 1987). The non-Mayan groups represent reference populations.

Geographic location of the population groups sampled in this study. Map illustrating the location of the reference populations. Map of Mesoamerica indicating the location of the Mayan groups
Collection of samples and DNA isolation
All samples were collected as whole blood in EDTA Vacutainer tubes from unrelated individuals traced back at least two generations. Samples were procured according to the ethical guidelines as indicated by Florida International University’s Institutional Review Board. Consent forms were distributed and subsequently explained in Spanish and in the corresponding Mayan dialect. The blood cells were lysed and leukocyte nuclei were separated from the rest of the blood components as previously reported (Antunez de Mayolo et al. 2002). DNA was then purified using proteinase-K digestion and standard organic phenol-chloroform extraction (Novick et al. 1995). All samples were stored at −80°C when not in use.
DNA amplification of PAI loci
PCR amplification of each PAI was performed as described previously (Novick et al. 1995) in 15-μl reactions containing 5 ng/μl of each primer and 200 μM dNTPs (Applied Biosystems) in 50 mM KCl, 1.5 mM MgCl2, 10 mM Tris–HCl, pH 8.3 and 1.0 U of Ampli-Taq DNA polymerase (Perkin Elmer). Samples were cycled in a standard three-step PCR profile with the parameters previously published elsewhere (Batzer et al. 1996; Antunez de Mayolo et al. 2002). Subsequent to DNA amplification, the samples were loaded into 3% agarose, 1X TAE gels. Amplicons were subjected to electrophoresis, visualized by staining with ethidium bromide and photographed under UV light. The primers used in this study were APO (Karathanasis 1985), ACE (Tiret et al. 1992), COL3A1 (Milewicz et al. 1996), D1 (Batzer et al. 1996), F-13 (Batzer et al. 1996), NBC4 (Roy et al. 1999), PR (Antunez de Mayolo et al. 1999), PV92 (Batzer et al. 1994), TCR (Rowen et al. 1996) and TPA25 (Batzer and Deininger 1991) (see Table 2).
Previously reported allelic frequencies
The allelic frequencies for the ten loci are reported here for the first time for the following populations: Cakchiquel (Maya group from Guatemala; number of samples, n=56); Quiche (Maya group from Guatemala; n=27); Benin (West African population; n=109).
Rwanda from East Africa was previously reported for the PR locus (Antunez de Mayolo et al. 1999). Five loci (ACE, APO, FXIIIB, PV92 and TPA25) were reported before for the following populations: Navajo (n=41), Maya from Buctzotz, Yucatan, Mexico (n=21), Maya from Campeche, Yucatan, Mexico (n=20) and Alaskan Eskimo (n=46) (Novick et al. 1998). Also, the same five loci mentioned above plus D1 were published before for the following populations: Madras from southeastern India (n=50) and Chinese (n=49) (Antunez de Mayolo 2002). The Galician population was examined previously for APO, COL3A1, PR, PV92 and TPA25 (Antunez de Mayolo et al. 1999; Terreros et al. 2005). The loci not mentioned for the above-indicated populations are reported in the present study for the first time.
Data analysis
Allelic and genotypic frequencies were estimated by the gene-counting method (Li 1976). Unbiased estimates of expected heterozygosity, its average per population and standard errors were calculated utilizing the BIOSYS-2 program (Black 1996). The unbiased estimate of expected heterozygosity was obtained as h=n (1−Σ p 2i )/n−1, where n is the number of chromosomes sampled and p is the frequency of the i allele (Nei 1987). An exact test using Levene’s correction for small sample sizes was employed to verify whether the observed heterozygosity frequencies agreed with those expected under the hypothesis of Hardy–Weinberg equilibrium. All calculations were done using the BIOSYS-2 program (Black 1996). The G test was performed to assess the genetic differences between members of all possible pairs of populations. This analysis is designed to ascertain if the genetic differences between all pairs of populations are statistically significant. In addition, a separate test to assess the significance of population heterogeneity was performed using the chi-square (Roff and Bentzen 1989) analysis for small numbers.
The level of intra- and inter-population variability (H s, G st and H t) was assessed using the DISPAN program (Ota 1993). Phylogenetic analyses based on the Alu frequencies were conducted using CONTML, a restricted Maximum-Likelihood (ML) routine in PHYLIP Version 3.5C (Felsentein 1993). Interior branch reliability was tested by means of 1,000 bootstrap replications using the SEQBOOT program. A Principal Component (PC) analysis (Sneath and Sokal 1973) based on the allelic frequency data was performed to infer the relationships among the groups using the NTSYSpc program (Rohlf 1998). The relative degree of gene flow experienced by each of the Mayan groups was estimated by plotting the distance from the centroid versus the heterozygosity (Harpending and Ward 1982). According to this analysis, under an island model of population structure, it is postulated that a simple linear relationship is expected between the heterozygosity of a population and the distance of that population from the centroid, which is defined as the arithmetic mean of the allele frequencies. Mathematically, this relationship is defined as: r i = (p i−P)2/(P)(1−P), where r i is the distance from the centroid, and p i and P are the frequency of the insertion in population i and in the four Mayan populations combined, respectively. The expectation is that populations that have experienced more gene flow and/or possess higher effective population size than average will fall above the theoretical prediction (regression line), while populations that have had less gene flow will fall below. A actual regression line was computed based on the observed heterozygosity values to ascertain if the populations examined comply with the premises of the centroid analysis. Hierarchical determination via the analysis of molecular variance (AMOVA), grouping populations according to geographic criteria (i.e., Mesoamerica, southwestern USA and Alaska) or linguistic affiliations (i.e., Quichean, Yucatec, Na-Dene and Eskimo-Aleut), was conducted using the Arlequin program (1,023 permutations) (Excoffier et al. 1992; Schneider et al. 2000). The AMOVA test was performed to ascertain any potential correlation between genetic diversity and geographic or linguistic partitioning.
Results
Intra-population diversity
Table 3 presents the Alu allelic frequencies as well as the heterozygosity levels of the populations examined. Additionally, genotypic distributions are given in Table 4. Although the aim of this study is not to focus on allelic frequencies of specific loci, certain tendencies should be noted since they directly relate to the central issue of this report. Among the four Mayan groups, four (APO, COL3A1, NBC4 and TCR) of the ten loci examined possess allelic frequencies at fixation for the insertion or lack of insertion state. Of these four loci, three (APO, NBC4 and PV92) represent fixation of the insertion allele. Similarly, among the other two Native American populations examined (Alaskan Eskimos and Navajo) in the present study, allelic fixation was detected in four of the ten loci (APO, COL3A1, F13 and TCR). In contrast, only two of the worldwide reference populations studied demonstrate fixation—Benin at PR and the Han Chinese at PR and TCR—for the absence of the insertion allele. None of the non-Native American populations exhibit fixation for the insertion allele. Also, contrary to the extreme frequencies at or near fixation seen in the majority of the loci among the Mayans and the other two Native American groups, the other worldwide populations are characterized, for the most part, by intermediate allelic frequencies (i.e., 0.2–0.8).
Allelic frequencies and probability values concerning Hardy–Weinberg equilibrium expectations are given in Table 3. Among the Mayan groups, observed heterozygosity for each population averaged across the ten loci range from 0.133 in the Quiche to 0.249 in the Buctzotz. The corresponding values for the other two Native American populations are 0.114 in the Navajo and 0.175 for the Alaskan Eskimo. The worldwide populations excluding the Mayan groups as well as the Navajo and Alaskan populations exhibit average observed heterozygosities across all loci ranging from 0.238 to 0.361, representing the Han Chinese and Benin, respectively. The highest average observed heterozygosities for all the loci were detected in the two African groups. The lowest observed heterozygosity values were detected among the Guatemalan Mayan groups (0.133 for the Quiche and 0.171 for the Cakchiquel) and the Navajo (0.114) (Table 3). Of the 130 tests, 10 revealed departures from Hardy–Weinberg equilibrium expectations, yet this value fell to 3 subsequent to Bonferroni corrections. Considering that at a 5% level of significance one would expect about seven of the population-locus permutations to fluctuate from Hardy–Weinberg equilibrium by chance alone, some of the 10 cases of disequilibria may not represent deviations due to factors other than chance. The three populations/loci in disequilibrium at the 0.005 level after Bonferroni corrections were D1 in Navajos and NBC4 and TCR in Galicia.
Inter-population diversity
Significant differences in average allelic frequencies were found between the Mayan groups and the other Native American populations (P<0.001), Mayan and European groups (P<0.001), Mayan and African populations (P<0.001), Mayans and Asians (P<0.001), Native Americans (other than Mayan) and Europeans (P<0.0001), Native Americans and Asians (P<0.0001), Native Americans and Africans (P<0.0001), Asians and Europeans (P<0.001), Asians and Africans (P<0.001) and Africans and Europeans (P<0.001) (Roff and Bentzen 1989). When the G test was performed, statistically significant genetic differences were observed in all pair-wise comparisons of groups except between the two Guatemalan Mayan populations from Cakchiquel and Quiche, and the Navajo in relation to the two Guatemalan groups (Table 5).
Genetic variance within populations (H s) and between populations (G st) was assessed (Table 6). The intra-population and inter-population components of genetic variance were examined for each locus (Table 6). G st is an estimation of the genetic differentiation among populations. For this study, the groups were segregated into three clusters: (1) the whole set of populations, (2) Native American populations excluding the Mayan groups and (3) the Mayan groups. Overall, the inter-population variation among the Mayan groups is 18% of the total (H t), while for the other Native Americans, G st is 20% of the total. As expected, most of the variance, therefore, is found within groups. The mean intra-population variance for all ten PAIs is highest for the whole set of populations (24%) and lowest for the Native Americans excluding the Mayan groups (21%). The Hs value for the Mayans is 22%. Except for three loci (D1, NBC4 and PV92), the Mayan groups exhibit higher intra-population variance than the other two Native American populations. The whole set of populations possesses higher intra-population variance than the Mayan groups in all but three of the markers (D1, PR and TPA).
In terms of inter-population variance, the average value for all ten loci is highest for the whole set of populations (30%) and the lowest for the Mayan groups (4%). The Native American populations exhibit a G st value comparable to the Mayan groups (5%). Six (APO, COL3A1, D1, NBC4, PR and TCR) of the ten loci possess higher inter-population variation in the Mayan groups in relation to the other two Native American populations. Only D1 in the Mayan groups and TPA in the Navajo and Alaskan-Eskimo exhibit G st values higher than the whole set of populations.
Phylogenetic and statistical analyses
To examine phylogenetic relationships among populations, the Mayan groups were examined in relation to several worldwide reference populations (Alaskan Eskimo, Navajo, Benin, Rwanda, China, Madras and Galicia) using ML analysis. The ML tree (Fig. 2) was rooted with a hypothetical ancestral population with the insertion frequencies set at zero for each of the ten loci (Batzer et al. 1994). Two clusters are observed in the ML dendrogram. Nine bifurcations segregate these 11 populations plus the hypothetical ancestor group to form the tree. Eight of the nine nodes are above 50%. In one cluster, the hypothetical ancestral population segregates in close proximity to the two sub-Saharan African groups, Benin and Rwanda. In this same branch, the two Caucasian populations from Galicia and Madras are located away from the sub-Saharan groups and closer to the other clade. In the second cluster, the Cakchiquel Maya and Quiche Maya, both from Guatemala, group as sister taxa. The two Mayan populations from the Yucatan, Campeche and Buctzotz, cluster within the same clade at some distance from the Guatemalan Mayan populations. The Alaskan Eskimos segregate in between the Mayan populations from the Yucatan, while the Navajo holds an intermediate position bisecting the Yucatan Mayan populations from the Guatemalan Mayan groups.

Maximum-likelihood tree illustrating human phylogenetic relationships. This tree was generated directly from the allelic frequencies of ten Alu insertion polymorphisms. Bootstrap values based on 1,000 replications are shown for each internal branch
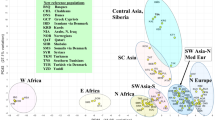
A PC plot was constructed with the four Mayan groups and the worldwide reference populations (Fig. 3). PC1 and PC2 represent 69.5 and 18.3%, respectively, of the total variance. PC1 separates the African populations from Asians, Mayans and the other Native American groups. The four Mayan groups segregate within the Native American/Asian cluster. The Mayan groups do not plot by themselves. The distance of the Mayans from the Yucatan (Buctzotz and Campeche) from the Mayan groups from Guatemala is equivalent to the one separating most of the Mayan populations from the Navajo and Alaskan Eskimo.

Principal component analysis derived from the allelic frequency of the ten Alu insertion polymorphisms from all 13 collections
A centroid analysis was performed to ascertain relative gene flow and/or differences in effective population size among the four Mayan groups (Fig. 4). The solid regression line represents the theoretical average values, while the dotted line is the actual regression line based on the observed heterozygosity instead of the expected. In this analysis, populations above the theoretical linear regression line exhibit greater-than-average gene flow and/or effective population size, while populations below the regression line represent groups with lower-than-average gene flow and/or effective population size. In the centroid test, the Buctzotz and the Campeche Mayan groups plot above the regression line, indicating higher than average gene flow and/or effective population size, while the Cakchiquel and Quiche segregate below the regression line, pointing to lower than average gene flow and/or effective population size. The Buctzotz and Quiche groups are outliers further out on either side of the regression line. For the purpose of assessing whether geographical and/or linguistic affiliations partition along the lines of genetic differences, the AMOVA test was performed. In essence, the AMOVA test ascertains whether there is a correlation or parallelism between genetic differentiation on one hand and geographical categories or linguistic types on the other. For this purpose, the comparison of genetics and geography was made by grouping the Mesoamerica populations from Cakchiquel, Quiche, Buczotcz and Campeche together into one group; the second group was assembled with the Eskimo and Navajo populations. In the case of linguistics, four groups were generated. The first is comprised by the two Mayan groups from Guatemala, the second is made up of the two groups from Yucatan, the Eskimos constitute the third and the Navajo the fourth. With these groupings, the AMOVA performed comparisons between populations within groups as well as among groups of populations. Of all of these comparisons, only linguistic and genetic differences correlated significantly at the level of among groups of populations. Comparisons between genetic partitioning in relation to geography and linguistic affiliations generated insignificant P-values among populations within groups (see Table 7). The analyses described above suggest that the Mayan groups examined may not represent a genetically homogeneous population.

The heterozygosity of each Mayan group is plotted against the distance of the population from the centroid
Discussion
Previous studies involving PAIs indicate higher insertion frequencies among Native American and Asian populations in comparison to African groups (Stoneking et al. 1997; Watkins et al. 2001). The allelic frequencies observed in the four Mayan groups examined in the present study corroborate these previous observations. Only the COL3A1 and TCR loci exhibit higher insertion frequencies in the African populations from Benin and Rwanda than in Native American, Asian and European groups. It is likely that the high allelic frequency levels at or near fixation in the Mayan and the other Native American groups is the result of genetic drift, bottle neck events and/or founder effects.
The Navajo display the lowest observed heterozygosity of all the populations examined. The second and third populations with the lowest observed heterozygosity are the Quiche and Cakchiquel Mayan groups from Guatemala. These low heterozygosity values most likely reflect reduced genetic diversity resulting from founder effect, genetic isolation and/or inbreeding. Relative to the Mayan groups from the Yucatan Peninsula, the two southern Guatemalan groups possess lower observed heterozygosity. The marked differences in observed heterozygosity between these two geographical regions may be related to higher levels of gene flow experienced by the Buctzotz and Campeche groups at the northern fringes of the Mayan range as compared to the Cakchiquel and Quiche at the southern extreme of their territory. Limited gene flow among the Guatemalan groups is corroborated by the position of these two groups below the regression line of the centroid analysis. The extensive commerce and interaction with technologically advanced neighboring northern tribes including the Aztecs and Toltecs could at least in part explain the greater observed heterozygosity of the Yucatan Mayans in relation to their southern counterparts with a limited number of trading partners. In addition, the inaccessibility of the mountainous terrain occupied by the Cakchiquel and Quiche as compared to the unstructured, easy-to-travel Yucatan plateau may have contributed to the relative genetic isolation of the Guatemalan groups.
The overall intra-population genetic diversity is highest for the worldwide set (24%) of populations compared to the Mayan groups (22%) and the two other Native American populations (21%). Overall inter-population diversity is also highest in the worldwide populations (30%), while the Native American and Mayan sets exhibit comparable much lower values (5 and 4%, respectively). The G st for the worldwide set of populations is higher than those reported for 17 proteins (10%) and 6 VNTR/STR (19%) loci (Bortolini et al. 1998). The G ST values for the Native American set are in the range of previously reported data from other genetic systems. Somewhat higher G st values were obtained when studying protein (9–10%) (Bortolini et al. 1998) and blood group/protein (8–17%) (Fagundes et al. 2002) polymorphisms in Amerindians. Both intra- and inter-population variability among the Mayan groups are similar to the ones observed for the other Native American populations in this study. These results may suggest that the genetic diversity and uniqueness of the Mayan groups is comparable to the values obtained for geographically distant and linguistically distinct (i.e., Na-Dene and Eskimo-Aleut) Native American populations. The G test demonstrated that except for the comparisons between the two Guatemalan Mayan groups, all pair-wise combinations involving the four Mayan groups exhibit significant genetic differences. The Navajo was found to be genetically similar to both of the Guatemalan Mayan groups. These data may be indicative of the lack of genetic homogeneity among the four Mayan groups, especially when comparing the Guatemalan to the Yucatecan groups. These results do not support a homogeneous Mayan population.
The loose segregation of the four Mayan collections among the Alaskan Eskimo and Navajo in the ML tree also argues for their genetic heterogeneity (Fig. 2). The Guatemalan Cakchiquel and Quiche populations group in close proximity with the geographically distant, Na-Dene-speaking Navajo, while the Eskimo-Aleut-speaking Alaskan Eskimos segregate in between the Campeche and Buctzotz Mayans from the Yucatan. The four Mayan populations failed to group together into a Mayan cluster. Instead, the Mayan populations are found scattered within the Native American clade.
The scattered distribution of these four Mayans collections within the Asian and Native American populations in the PC plot (Fig. 3) likewise suggests the lack of an intimate genetic affinity among these Mayan groups. The expectations based on strong genetic relationships are a tight agglutination of the four Mayan groups away from the rest of the Asian/Native American populations, which is not observed. Although the genetic relationships reflected in the PC graph are not the same as in the ML dendrogram, in both analyses the Mayan groups often exhibit greater similarity to non-Mayan populations than to themselves. As expected, the populations from West and East Africa, traditionally exhibiting greater genetic diversity, segregate somewhat distantly from each other in the upper left-hand quadrant of the plot, while the Galicians map in the right lower portion of the graph away from the African and Asian/Native American clusters. The population from Madras segregates intermediately between the Galician and the Asian/Native American cluster, but considerably closer to the latter, probably reflecting some Asiatic influence.
The overall AMOVA using all ten loci was performed to determine if genetic variability correlates with geographic and/or linguistic partitioning. Significant parallelism was detected along linguistic lines when groups of populations were compared. These results may indicate the existence of significative genetic differences between the linguistically distinct northern Mayan groups in the Yucatan peninsula (Yucatec language subfamily) and the southern Guatemalan Mayans (Cakchiquel and Quiche; Quichean language subfamily). These data also underscore the genetic distinctness of the Guatemala and the Yucatan Mayan groups.
The centroid test provides information on the effective population size and/or relative genetic flow among the groups being analyzed (Harpending and Ward 1982). In the plot, groups above the regression line possess a higher-than-average effective population size and/or experience greater gene flow. The opposite is expected of groups below the linear regression. Since the centroid test assumes an island model of population structure involving gene flow among groups, this analysis is particularly useful in the assessment of relative gene flow among groups of comparable size and capable of interbreeding due to their geographical proximity. Considering that the four Mayan groups under investigation are of comparable size, their segregation within the centroid plot should be indicative of gene flow. In order to assess how suitable the assumptions of the centroid model of population dynamics are for the Mayan groups examined in this study, actual and theoretical regression lines were generated using the observed and expected heterozygosity values, respectively. The almost identical slopes of both lines indicate that the Mayan groups examined do not violate the premises of the centroid analysis. In the present study, it is interesting that the two Mayan groups from the Yucatan peninsula segregate above the regression lines, suggesting greater-than-average gene flow for these ten PAI markers. On the other hand, the Cakchiquel and Quiche from southern Guatemala map below the line, indicating lower-than-average gene flow. Historical events and different degrees of geographical isolation may provide explanations for the greater levels of gene flow exhibited by the two peninsular groups as compared to the Cakchiquel and Quiche at the southern range of the Mayan empire. Historically, northern Mayan groups interacted with people to the north such as the Aztec. In addition, the geographical proximity and the flat accessible terrain of the Yucatan plateau provided for extensive contact with technologically sophisticated populations to the north such as the Toltecs. In contrast, the two southern Mayan groups occupy less accessible mountainous regions that may have contributed to greater geographical isolation. In addition, the Cakchiquel and the Quiche may not have been involved in extensive interaction with the relatively less-advanced neighbors to the south. The greater levels of heterozygosity exhibited by the two northern Mayan groups may suggest a longitudinal divide among Mayan groups. It also suggests a lack of genetic uniformity among the Mayans. Our findings, although thought-provoking, need to be tempered and considered preliminary until additional Mayan and other Native American populations as well as greater numbers of individuals per population are examined.
In conclusion, a number of parameters of population genetic interest were assessed in the present study. Overall, the data generated suggest that genetic heterogeneity exists among the four Mayan collections examined. The Mayan groups sampled represent two from the northern and two from the southern range of their historical empire. These four collections encompass the Cakchiquel and Quiche from the traditional mountainous Mayan homeland as well as the Buctzotz and Campeche at the northern fringes of their empire in the Yucatan plateau. These four Mayan groups exhibit extreme allelic frequencies at or close to fixation for the insertion or lack of insertion allele. Except for one pair test, the G test for genetic homogeneity among populations indicates that the Mayan groups are statistically different from each other. Also, the inter-population variation among the four Mayan groups is comparable to that of non-Mayan Native American populations, corroborating similar levels of heterogeneity. Phylogenetic analyses in the form of a ML tree and a PC plot indicate genetic affinities among the four Mayan groups comparable to geographically distant Native American populations. In addition, the AMOVA test demonstrated that genetic diversity correlated to linguistic partitioning when groups of populations were compared. Since two of the groups of populations studied included two distinct Mayan language families, these results also suggest intra-Mayan subpopulation structure along linguistic lines. Furthermore, the Buctzotz and Campeche groups from the north seem to have experienced greater gene flow in comparison to the Guatemalan Mayans. However, the significance of our findings needs to be tempered and considered preliminary due to the paucity of the Mayan and other Native American populations as well as individuals examined in this study.
References
Antunez De Mayolo A, Antunez De Mayolo G, Thomas E, Reategui EP, Brown M, Herrera RJ (1999) Worldwide distribution of a polymorphic Alu insertion in the progesterone gene. In: Papiha SS, Deka R, Chakraborty R (eds) Genomic diversity: applications in human population genetics. Kluwer Academic/Plenum Publishers, New York, pp 213–222
Antunez De Mayolo G, Antunez De Mayolo P, Antunez De Mayolo A, Papiha Ss, Hammer MF, Yunis E, Yunis J, Damodaran C, York D, Martinez De Pancorbo M, Stoneking M, Caeiro JL, Puzyver VP, Herrera RJ (2002) Phylogenetics of worldwide human populations as determined by polymorphic Alu insertions. Electrophoresis 23:3346–3356
Arnaiz-Villena A, Vargas-Alarcón G, Granados J, Gómez-Casado E, Longas J, Gonzales-Hevilla M, Zuñiga J, Salgado N, Hernández-Pacheco G, Guillen J, Martinez-Laso J (2000) HLA genes in Mexican Mazatecans, the peopling of the Americas and the uniqueness of Amerindians. Tissue Antigens 56:405–416
Batzer MA, Deininger PL (1991) A human-specific subfamily of Alu sequences. Genomics 9:481–487
Batzer MA, Stoneking M, Alegria-Hartman M, Bazan H, Kass DH, Shaikh TH, Novick GE, Ioannou PA, Scheer WD, Herrera RJ, Deininger PL (1994) African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci USA 91:12288–12292
Batzer MA, Arcot SS, Phinney JW, Alegria-Hartman M, Kass DH, Milligan SM, Kimpton C, Gill P, Hochmeister M, Ioannou PA, Herrera RJ, Boudreau DA, Scheer WD, keats BJB, Deininger PL, Stoneking M (1996) Genetic variation of recent Alu insertions in human populations. J Mol Evol 42:22–29
Bianchi NO, Bailliet G, Bravi C, Carnese RF, Rothhammer F, Martinez-Marignac VL, Pena SDJ (1997) Origin of Amerindian Y-chromosomes as inferred by the analysis of six polymorphic markers. Am J Phys Anthropol 102:79–89
Bianchi NO, Catanesi CI, Bailliet G, Martinez-Marignac VL, Bravi CM, Vidal-Rioja LB, Herrera RJ, López-Camelo JS (1998) Characterization of ancestral and derived Y-chromosome haplotypes of New World populations. Am J Hum Genet 63:1862–1871
Black FL (1991) Reasons for failure of genetic classifications of South Amerindian populations. Hum Biol 63:763–774
Black W (1996) BIOSYS-2: a computer program for the analysis of allelic variation in genetics. Department of Microbiology, Colorado State University. Available at ftp://www.lamar.colostate.edu/pub/wcb4/
Bonatto SL, Salzano FM (1997) A single and early migration for the peopling of the Americas supported by mitochondrial DNA sequence data. Proc Natl Acad Sci USA 94:1866–1871
Bortolini MC, Baptista C, Callegari-Jacques SM, Weimer TA, Salzano FM (1998) Diversity in protein, nuclear DNA and mtDNA in South Amerinds. Agreement or discrepancy. Ann Hum Genet 62:133–145
Carroll ML, Roy-Engel AM, Nguyen SV, Salem AH, Vogel E, Bethaney V, Myers J, Ahmad Z, Nguyen L, Sammarco M, Watkins WS, Henke J, Makalowski W, Jorde LB, Deininger PL, Batzer M (2001) Large-scale analysis of the Alu Ya5 and Ya8 subfamilies and their contribution to human genomic diversity. J Mol Biol 311:17–40
Coe MD (1992) The history of the Maya decipherment in breaking the Maya code. Thames & Hudson, Inc. Publishers, London, New York
Crawford MH (1998) The origins of Native Americans. Evidence from anthropological genetics. Cambridge University Press, Cambridge
Donaldson CJ, Crapanzano JP, Watson JC, Levine EA, Batzer MA (2002) PROGINS Alu insertion and human genomic diversity. Mutat Res 501:137–141
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA data. Genetics 131:479–491
Fagundes NJR, Bonatto SL, Callegari-Jacques SM, Salzano FM (2002) Genetic, geographic, and linguistic variation among South American Indians: possible sex influence. Am J Phys Anthropol 117:68–78
Felsentein J (1993) PHYLIP (Phylogeny Inference Package), version b 3.5. Department of Genetics, University of Washington, Seattle
Greenberg JH (1987) Language in the Americas. Stanford University Press, Stanford
Harpending HC, Ward RH (1982) Chemical systematic and human population. In: Nitecki M (ed) Biochemical aspects of evolutionary biology. University of Chicago Press, Chicago, pp 213–252
Hillis DM (1999) SINEs of the perfect character. Proc Natl Acad Sci USA 96:9979–9981
Karathanasis SK (1985) Apolipoprotein multigene family: tandem organization of apolipoprotein AI, CIII, and AIV genes. Proc Natl Acad Sci USA 82:6374–6378
Li CC (1976) First course in population genetics. Boxwood Press, Pacific Groove
Martinez L, Reategui EP, Fonseca L, Sierra-Montes JM, Terreros MC, Pereira-Simon S, Herrera RJ (2005) Superimposing Polymorphism: the case of a point mutation within a polymorphic Alu insertion. Hum Hered 59:109–117
Mateus-Pereira LH, Socorro A, Fernandez I, Masleh M, Vidal D, Bianchi NO, Bonatto SL, Salzano FM, Herrera RJ (2005) Phylogenetic information in polymorphic L1 and Alu insertions from East Asians and Native American populations. Am J Phys Anthropol 128:171–184
Milewicz DM, Byers PH, Reveille J, Hughes J, Duvic M (1996) A dimorphic Alu Sb-like insertion in COL3A1 is ethnic-specific. J Mol Evol 42:117–123
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Novick GE, Novick CC, Yunis E et al (1995) Polymorphic human-specific Alu insertions as markers for human identification. Electrophoresis 16:1596–1601
Novick GE, Novick CC, Yunis J, Yunis E, Antunez De Mayolo P, Scheer WD, Deininger P, Stoneking M, York D, Batzer MA, Herrera RJ (1998) Polymorphic Alu insertions and the Asian origin of Native American populations. Hum Biol 70:23–39
Ota T (1993) DISPAN: genetic distance and phylogenetic analysis. Institute of Molecular Evolutionary Genetics, Pennsylvania State University. Available at ftp://www.bio.indiana.edu/molbio/ibmpc/
Roff DA, Bentzen P (1989) The statistical analysis of mitochondrial DNA polymorphism: χ2 and the problem of small samples. Mol Biol Evol 6:539–545
Rohlf FJ (1998) NTSYSpc numerical taxonomy and multivariate analysis system, version 2.02i. Applied Biostatistics Inc., Department of Ecology and Evolution, State University of New York
Rowen L, Koop B, Hood L (1996) The complete 685-Kilobase DNA sequence of the human βT cell receptor locus. Sciences 272:1755–1765
Roy AM, Caroll ML, Kass DH, Nguyen SV, Salem A, Batzer MA, Deininger P (1999) Recently integrated human Alu repeats: finding needles in the haystack. Gentetica 107:149–161
Salzano FM (2002) Molecular variability in Amerindians: widespread but uneven information. An Acad Bras Ciênc 74:223–263
Salzano FM, Callegari-Jacques SM (1988) South American Indians: a case study in evolution. Clarendon Press, Oxford
Santos FR, Delfín LR, Pena SDJ, Moore J, Weiss KM (1996) North and South Amerindians may have the same major founder Y chromosome haplotype. Am J Hum Genet 58:1369–1370
Schneider S, Roessli D, Excoffier L (2000) Arlequin ver. 2.000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland
Silva WA, Bonatto SL, Holanda AJ, Ribeiro-dos-Santos AK, Paixão BM, Goldman GH, Abe-Sandes K, Rodriguez-Delfin L, Barbosa M, Paçó-Larson ML, Petzl-Erler ML, Valente V, Santos SEB and Zago MA (2002) Mitochondrial genome diversity of Native Americans supports a single early entry of founder populations into America. Am J Hum Genet 71:187–192
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. Freeman, San Francisco
Stoneking M, Fontius J, Clifford S, Soodyall H, Arcot SS, Saha N, Jenkins T, Tahir MA, Deininger PL, Batzer MA (1997) Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res 7:1061–1071
Terreros MC, Martinez L, Herrera RJ (2005) Polymorphic Alu insertions and genetic diversity among African populations. Hum Biol 77:675–704
Tiret L, Riget B, Visvikis S, Breda C, Corvol P, Cambien F, Soubrier F (1992) Evidence from combined segregation and linkage analysis, that a variant of the angiotensin I-converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum Genet 51:197–205
Watkins WS, Ricker CE, Bamshad MJ, Carroll ML, Nguyen SV, Batzer MA, Harpending HC, Rogers AR, Jorde LB (2001) Patterns of ancestral human diversity: an analysis of Alu-insertion and restriction-site polymorphisms. Am J Hum Genet 68:738–752
York DS, Blum VM, Low JA, Rowold D, Puzyrev V, Saliukov V, Odinokova O, Herrera RJ (1999) Phylogenetic signals from point mutations and polymorphic Alu insertions. Genetica 107:163–170
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Herrera, R.J., Rojas, D.P. & Terreros, M.C. Polymorphic Alu insertions among Mayan populations. J Hum Genet 52, 129–142 (2007). https://doi.org/10.1007/s10038-006-0089-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0089-y
Keywords
This article is cited by
-
Frequency of Alu insertions within the ACE and PR loci in Northwestern Mexicans
BMC Research Notes (2017)
-
Genomic insights on the ethno-history of the Maya and the ‘Ladinos’ from Guatemala
BMC Genomics (2015)
-
Frequency of thiopurine S-methyltransferase mutant alleles in indigenous and admixed Guatemalan patients with acute lymphoblastic leukemia
Medical Oncology (2013)
