
Abstract
Type 2 diabetes mellitus (T2DM) is a group of multifactorial disorders due to either defective insulin secretion or action. Despite the fact that numerous genetic researches of T2DM have been pursued, the pathogenic mechanisms remain obscure. We encountered a T2DM family associated with a balanced reciprocal translocation, t(3;9)(p21.31;q33.1). To isolate a candidate gene susceptible to T2DM, we constructed physical maps covering both the 3p and 9q breakpoints of the translocation in the family. Consequently, the inositol hexaphosphate kinase 1 gene (IHPK1) (OMIM *606991) was found to be disrupted at the 3p21.31 breakpoint. We then carried out sequence analysis for all coding regions of IHPK1 in 405 unrelated T2DM patients in order to validate whether aberrations of the gene are common in T2DM patients, but we failed to detect any pathogenic changes. The disruption of IHPK1 or another predisposing gene affected by position effect of the translocation may explain the T2DM phenotype at least in this family. Alternatively, the IHPK1 disruption in the family is a chance association.
Similar content being viewed by others


Introduction
Type 2 diabetes mellitus (T2DM, MIM 125853), previously referred to as non-insulin-dependent diabetes mellitus (NIDDM), is a common metabolic disorder characterized by chronic hyperglycemia due to either defective insulin secretion or action. It is generally accepted that T2DM is a group of multifactorial diseases caused by both genetic and environmental factors. Possible associations have been reported between T2DM and mutation(s) in either of the following genes: GPD2 at 2q24.1 (Novials et al. 1997), NEUROD1 at 2q32 (Malecki et al. 1999), IRS1 at 2q36 (Laakso et al. 1994), CAPN10 at 2q37.3 (Horikawa et al. 2000), PPARG at 3p25 (Deeb et al. 1998), SLC2A2 at 3q26.1–q26.3 (Tanizawa et al. 1994), MAPK8IP1 at 11p12–p11.2 (Waeber et al. 2000), TCF1 at 12q24.2 (Triggs-Raine et al. 2002), SLC2A4 at 17p13 (Kusari et al. 1991), TCF2 at 17qcen–q21.3 (Furuta et al. 2002), or HNF4A at 20q12–q13.1 (Hani et al. 1998). Toward the identification of novel genes susceptible to T2DM and better understanding of the pathogenesis of T2DM, genome-wide scanning of affected pedigrees with an inheritance similar to Mendelian pattern or breakpoint analyses of chromosomal rearrangements associated with T2DM may be useful.
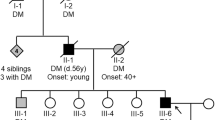
Recently, we encountered a Japanese girl with a balanced chromosomal translocation, t(3;9)(p21.31;q33.1), and T2DM. Her mother also had T2DM and the translocation. We hypothesized that a susceptible gene to T2DM is disrupted at either of breakpoints at 3p21.31 or 9q33.1. Here, we present data of breakpoint characterization and mutation analysis.
Materials and methods
The family examined
The proband, a 14-year-old Japanese girl, was diagnosed to have T2DM at age of 6 years after glycosuria was initially pointed out. She had been severely obese since her infancy, and body mass index (BMI) at diagnosis was 31.4 kg/m2 (weight 50.7 kg and height 127 cm). Although she required neither insulin injections nor an oral antidiabetic agent, she received repeated dietary and exercise treatments. Her maximum fasting plasma glucose and HbA1c levels before treatments were 202 mg/dl (normal value: <110 mg/dl) and 13.9% (normal value: 4.0–6.5%), respectively. After the latest treatment, HbA1c was 5.9%. GTG-banding and fluorescence in situ hybridization (FISH) analyses were performed for ruling out the Prader–Willi syndrome. The initial karyotype was reported as 46,XX,t(3;9)(p14.1;q31).ish 15q11.2(SNRPNx2). Further FISH analyses later localized the precise breakpoints of both derivative chromosomes to 3p21.31 and 9q33.1. Thus, her karyotype was revised as 46,XX,t(3;9)(p21.31;q33.1).
Her 47-year-old obese mother was also diagnosed as T2DM after her second pregnancy, and her T2DM is currently under control with an oral antidiabetic agent. A 15-year-old elder brother of the proband showed mild obesity but no apparent hyperglycemia. The mother and elder brother both had the same balanced translocation, but her healthy father had a normal karyotype. Blood samples of her maternal grandparents and three other siblings of the mother were not available, but none of them had T2DM.
Fluorescence in situ hybridization (FISH)
FISH analysis using bacterial artificial chromosome (BAC) or cosmid clones was performed on metaphase chromosomes of the proband and of a normal control. Chromosome preparations made from lymphoblastoid cell lines were baked at 75°C for 2 h, denatured in 70% formamide/2× SSC at 70°C for 2 min, and then dehydrated in 70 and 100% ethanol. Probe DNAs were labeled with SpectrumOrange-11-dUTP or SpectrumGreen-11-dUTP (Vysis, Downers Grove, IL, USA) by nick translation and denatured at 70°C for 10 min. Probe-hybridization mixture (15 μl) was applied on the chromosome slides and incubated at 37°C for 16–72 h. Slides were washed at 43°C in 50% formamide/2× SSC, 2× SSC, 1× SSC, 0.1% Triton-X-100/4× SSC, twice of 4× SSC, and 2× SSC, respectively, before mounting in antifade solution (Vector, Burlingame, NY, USA) containing DAPI. Fluorescence photomicroscopy was performed under a Zeiss Axioskop microscope equipped with a quad filter set with single-band excitation filters (84000, Chroma Technology Corporation, Brattleboro, VT, USA). Images were collected and merged with a cooled CCD camera (TEA/CCD-1317-G1, Princeton Instruments, Trenton, NJ, USA) and IPLab/MAC software (Scanalytics, Fairfax, VA, USA).
Construction of physical maps and screening of clones spanning the 3p21.31 and 9q33.1 translocation breakpoints
The RPCI-11 human BAC library and the CalTech human BAC library were used to collect clones mapped around 3p21.31 or 9q33.1. BAC/cosmid contigs were constructed using either electronic or actual PCR-based STS content mapping method, and clones incorporated to the contigs were used for subsequent FISH studies on the proband’s and normal person’s chromosomes. After isolation of a BAC clone spanning the 3p21.31 breakpoint, a cosmid library was prepared from the BAC DNA. Purified BAC DNA was isolated using Qiagen Midi-Prep columns (Qiagen, Chatsworth, CA, USA), partially digested with Sau3AI, and ligated to SuperCos1 cosmid vector according to the manufacturer’s instruction (Stratagene, La Jolla, CA, USA). Cosmid DNA was extracted with Qiagen Mini-Prep columns (Qiagen), and 120 cosmid clones were screened.
DNA sequencing of cosmid clones
Sequencing reactions were performed for 50 cycles (BAC and cosmid) or 25 cycles (PCR product) at 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min with BigDye Terminator ver. 3.0 Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA). The products were analyzed on an ABI 3100 automated DNA sequencer (Applied Biosystems) with the sequence analysis software (Applied Biosystems). Complete sequencing of a cosmid spanning the 3p21.31 breakpoint was performed by primer-walking strategy. T7 and SP6 primers for BAC and T7 and T3 for cosmid were used for clone-end sequencing.
Mutation analysis of genes at or around the 3p21.31 breakpoint
As for mutation screening of candidate genes, 405 unrelated, karyotypically normal Japanese patients with T2DM were analyzed. Peripheral blood samples were collected after obtaining informed consent, and genomic DNA was extracted according to the standard method. Seven primer sets were designed to cover the whole coding region as well as exon–intron boundaries of the genes (Table 1). PCR was cycled 35 times at 95°C for 30 s, 60°C (or 64°C) for 30 s, 72°C for 1 min in a volume of 20 μl, containing 1× PCR buffer with 2 mM MgCl2, 0.2 mM each dNTP, 1 μM each primer and 2.5 U Taq polymerase. PCR products were purified with ExoSAP-IT (Amersham Biosciences, Piscataway, NJ, USA) and directly sequenced. AutoAssembler ver. 2.1.1 software (Applied Biosystems) was used for sequence comparison. All protocols were approved by the Committee for the Ethical Issues on Human Genome and Gene Analysis, Nagasaki University.
Computational methods
Primer3 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi) was used to generate new STSs from clone-end sequences after the repetitive sequences were excluded with RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker). BLASTN searches (http://www.ncbi.nlm.nih.gov/BLAST/) were performed in GenBank + EMBL + DDBJ sequence databases (nr) and high-throughput genomic sequences (htgs) to obtain draft/complete sequences of overlapping clones. The Marshfield genetic map (http://research.marshfieldclinic.org/genetics/) and the UCSC Web site (http://genome.ucsc.edu/) were used for BAC-contig construction. The candidate genes around the breakpoint were detected with HGMP-RC NIX Session (http://www.hgmp.mrc.ac.uk/Registered/Webapp/nix/).
Results
Isolation of BAC clones spanning the breakpoints
We constructed two physical maps each covering the 3p21.31 or 9q33.1 breakpoint and the isolated BAC clones spanning them. Among BAC clones mapped around 3p21.31, RP11-78O10 (GenBank accession no. AC099330) and RP11-493K9 (AC104450) gave unique FISH signals on the derivative [der(3)] and normal chromosomes 3, while RP11-901L6 (AC099668), RP11-949J7 (AC104452), and RP11-3B7 (AC121247) gave signals on the der(9) and normal chromosomes 3. Only CTD-3126O21, of which BAC-end sequence information was available (Sp6: AQ148307, T7: AQ187080) and which are located between RP11-78O10 and RP11-901L6, was found to have signals on both the der(3) and der(9) in addition to normal chromosome 3 (data not shown). Regarding the 9q33.1 region, RP11-451E16 (AL365195), RP11-349E4 (AL355592), and RP11-98E22 (AL157780) gave FISH signals on the der(3) and normal chromosomes 9, while RP11-280M4 (AL358792) gave FISH signals on the der(9) and normal 9. RP11-564A4 (AL158831) located between RP11-451E16 and RP11-280M4 had signals on both der(3) and der(9) in addition to normal 9 (data not shown). Thus, CTD-3126O21 and RP11-564A4 span the 3p21.31 and 9q33.1 breakpoints, respectively.
Identification of cosmid-112 spanning the 3p21.31 breakpoint
Computational search around the 9q33.1 region using the complete sequence data of RP11-564A4 found no known genes. We therefore focused on the 3p21.31 breakpoint region. CTD-3126O21 was subcloned to cosmids, and a cosmid contig was constructed by PCR-based STS content mapping (Fig. 1). Nine new STSs (c19T3, c18T7, c61T3, c112T7, c51T3, c93T7, c12T3, c78T7, and c112T3) were generated from end-sequences of cosmids. FISH analysis showed seven cosmids were mapped centromeric to the 3p21.31 breakpoint, while two cosmids were telomeric to it. Finally, we identified a cosmid (cosmid-112) that showed signals on der(3), der(9), and normal chromosomes 3, indicating that the clone spanned the 3p21.31 breakpoint (Fig. 2). The complete 43,840-bp sequence of the cosmid was then determined.

Schematic representation of a BAC/cosmid physical map around the 3p21.31 breakpoint. Relative location of BAC clones used for fluorescence in situ hybridization (FISH) study is depicted in the upper part. The lower part shows a contig spanning the breakpoint. Normal and thick dotted vertical lines show positions of newly generated STS markers and the 3p21.31 breakpoint, respectively. Thick horizontal line depicts a BAC or cosmid clone spanning the breakpoint. Arrows and filled boxes indicate the transcriptional direction of genes and exons, respectively

Fluorescence in situ hybridization (FISH) analysis on the patient’s chromosomes using cosmid-112 spanning the breakpoint, a 3q subtelomere BAC clone (GS-56H22), and a 9p subtelomere BAC clone (GS-43N6). Arrows and arrowheads indicate signals of GS-56H22 and GS-43N6, respectively. Asterisk depicts split-signals of cosmid-112 on der(3) and der(9) chromosomes
Identification of the KIAA0263 gene at the 3p21.31 breakpoint
We searched for genes corresponding to cosmid-112 using HGMP-RC NIX Session (http://www.hgmp.mrc.ac.uk/Registered/Webapp/nix/) (Fig. 1). Complete sequences of two BACs [RP13-1056D16 (GenBank accession no. AC139451) and RP11-480C15 (GenBank accession no. AC141000)] overlapping with cosmid-112 were also analyzed (Fig. 1). Consequently, the IHPK1, (GenBank accession no. NM(153273), also known as the KIAA0263 gene (GenBank accession no. D87452), was identified to be disrupted at its intron 1 by the translocation. Two genes, GMPPB (GenBank accession no. NM(01334) and LOC375343 (GenBank accession no. XM(351552), were found at a region 19 kb-telomeric to the STS, c112T3, and at another region 16 kb-centromeric to c112T7, respectively (Fig. 1).
No pathogenic mutation of IHPK1, GMPPB, and LOC375343 among unrelated T2DM patients
Mutation analysis of IHPK1 performed in 405 unrelated T2DM patients identified nine different nucleotide changes, including four synonymous substitutions (c.351C→T, c.1041G→A, c.1127C→G, and c.1239C→T), one missense substitution (c.1057C→T: p.353R>W), three intronic substitutions (IVS1+118T→C, IVS1-29T→C, and IVS5+53G→C), and one intronic deletion (IVS5-26delG) (Table 2). All changes but the three synonymous substitutions (c.351C→T, c.1041G→A, and c.1239C→T) were also detected in normal controls, and allele frequencies of the changes were not significantly different between patients and controls (Table 2). Therefore, we concluded that they were not pathogenic. Additionally, we analyzed two other adjacent genes, GMPPB and LOC375343, in 192 unrelated T2DM patients, neither could we detect any abnormal changes among them (data not shown).
Discussion
To our knowledge, the family we studied is the third case of a chromosomal translocation associated with diabetes. A de novo unbalanced translocation t(X;10)(q22;p11) in a UK Caucasian female with adolescent-onset T2DM was previously reported (Owen et al. 2003). In another UK report, HNF4A at 20q12 was disrupted by a balanced translocation t(3;20)(p21.2;q12) in a MODY (maturity onset of diabetes of the young) family (Gloyn et al. 2002). Although these DM-associated chromosomal rearrangements are very rare, they may become a clue to identify a DM susceptible gene.
We have found that in our T2DM index patient, IHPK1 was disrupted at the 3p21.31 breakpoint of the translocation. This made the precise mapping of IHPK1 to 3p21.31. The breakage of the gene at intron 1 may have impaired its transcription. Nevertheless, we found no pathogenic mutations in the gene among other unrelated T2DM patients. As our analysis was focused on the entire coding region and exon–intron boundaries, changes at other regions, e.g., the introns or promoter, might have been overlooked. Alternatively, the disruption of IHPK1 in our family is a chance association.
IHPK1 (KIAA0263) has a 1323-bp open reading frame, consists of six exons, and encodes IHPK1 of 441 amino acids (Nagase et al. 1996). Its cDNA clone was originally obtained from a human immature myeloid cell line (KG-1). It is expressed in various tissues including testis, brain, heart, pancreas, liver, kidney, and skeletal muscle (Nagase et al. 1996) (Fig. 1). IHPK1 has been characterized by comparison with the mouse Ihpk1 sequence (Saiardi et al. 1999). Ihpk1-transfected cells showed inositol hexakisphosphate (InsP6) kinase activity. Purified InsP6 kinase has ATP synthase activity to change ADP to ATP using a phosphate from diphosphoinositol pentakisphosphate (PP-InsP5) (Voglmaier et al. 1996). This may indicate that human IHPK1 functions as energy reserves in intracellular sites and suggests its important role in cellular glycometabolism.
Position effect onto a certain gene by the translocation has also to be considered for the pathogenesis of T2DM in our index patient. Such an effect has been strongly implicated in several genetic diseases (Kleinjan and van Heyningen 1998), e.g., in patients with Rieger syndrome type 1, three different translocation breakpoints were identified within 90 kb upstream of the causative PITX2 gene (Datson et al. 1996; Semina et al. 1996; Flomen et al. 1998). If either of two genes, GMPPB and LOC375343, located close to the 3p21.31 breakpoint in our index case was affected by a position effect of the translocation, mutations in either gene would be present in a subset of unrelated T2DM patients. Under this assumption, we analyzed 192 unrelated T2DM patients but never detected any pathogenetic mutations. A few genome-wide scannings for T2DM in the Japanese population were performed but gave no evidence for linkage to the 3p21.31 region (Mori et al. 2002; Iwasaki et al. 2003). These data indicate no contribution or an extremely small contribution, even if present, of the locus at 3p21.31 to Japanese T2DM.
In conclusion, we found IHPK1 disruption in T2DM patients with familial t(3;9)(p21.31;q33.1). It remains to be seen how the abnormality in IHPK1 contributed to T2DM in this family by further functional analysis.
References
Datson NA, Semina E, van Staalduinen AA, Dauwerse HG, Meershoek EJ, Heus JJ, Frants RR, den Dunnen JT, Murray JC, van Ommen GJ (1996) Closing in on the Rieger syndrome gene on 4q25: mapping translocation breakpoints within a 50-kb region. Am J Hum Genet 59:1297–1305
Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J (1998) A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet 20:284–287
Flomen RH, Vatcheva R, Gorman PA, Baptista PR, Groet J, Barisic I, Ligutic I, Nizetic D (1998) Construction and analysis of a sequence-ready map in 4q25: Rieger syndrome can be caused by haploinsufficiency of RIEG, but also by chromosome breaks approximately 90 kb upstream of this gene. Genomics 47:409–413
Furuta H, Furuta M, Sanke T, Ekawa K, Hanabusa T, Nishi M, Sasaki H, Nanjo K (2002) Nonsense and missense mutations in the human hepatocyte nuclear factor-1 beta gene (TCF2) and their relation to type 2 diabetes in Japanese. J Clin Endocrinol Metab 87:3859–3863
Gloyn AL, Ellard S, Shepherd M, Howell RT, Parry EM, Jefferson A, Levy ER, Hattersley AT (2002) Maturity-onset diabetes of the young caused by a balanced translocation where the 20q12 break point results in disruption upstream of the coding region of hepatocyte nuclear factor-4alpha (HNF4A) gene. Diabetes 51:2329–2333
Hani EH, Suaud L, Boutin P, Chevre JC, Durand E, Philippi A, Demenais F, Vionnet N, Furuta H, Velho G, Bell GI, Laine B, Froguel P (1998) A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J Clin Invest 101:521–526
Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175
Iwasaki N, Cox NJ, Wang YQ, Schwarz PE, Bell GI, Honda M, Imura M, Ogata M, Saito M, Kamatani N, Iwamoto Y (2003) Mapping genes influencing type 2 diabetes risk and BMI in Japanese subjects. Diabetes 52:209–213
Kleinjan DJ, van Heyningen V (1998) Position effect in human genetic disease. Hum Mol Genet 7:1611–1618
Kusari J, Verma US, Buse JB, Henry RR, Olefsky JM (1991) Analysis of the gene sequences of the insulin receptor and the insulin-sensitive glucose transporter (GLUT-4) in patients with common-type non-insulin-dependent diabetes mellitus. J Clin Invest 88:1323–1330
Laakso M, Malkki M, Kekalainen P, Kuusisto J, Deeb SS (1994) Insulin receptor substrate-1 variants in non-insulin-dependent diabetes. J Clin Invest 94:1141–1146
Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, Saad M, Warram JH, Montminy M, Krolewski AS (1999) Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 23:323–328
Mori Y, Otabe S, Dina C, Yasuda K, Populaire C, Lecoeur C, Vatin V, Durand E, Hara K, Okada T, Tobe K, Boutin P, Kadowaki T, Froguel P (2002) Genome-wide search for type 2 diabetes in Japanese affected sib-pairs confirms susceptibility genes on 3q, 15q, and 20q and identifies two new candidate Loci on 7p and 11p. Diabetes 51:1247–1255
Nagase T, Seki N, Ishikawa K, Ohira M, Kawarabayasi Y, Ohara O, Tanaka A, Kotani H, Miyajima N, Nomura N (1996) Prediction of the coding sequences of unidentified human genes. VI. The coding sequences of 80 new genes (KIAA0201–KIAA0280) deduced by analysis of cDNA clones from cell line KG-1 and brain. DNA Res 3:321–329, 341–354
Novials A, Vidal J, Franco C, Ribera F, Sener A, Malaisse WJ, Gomis R (1997) Mutation in the calcium-binding domain of the mitochondrial glycerophosphate dehydrogenase gene in a family of diabetic subjects. Biochem Biophys Res Commun 231:570–572
Owen KR, Roland J, Smith K, Hattersley AT (2003) Adolescent onset Type 2 diabetes in a non-obese Caucasian patient with an unbalanced translocation. Diabet Med 20:483–485
Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH (1999) Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol 9:1323–1326
Semina EV, Datson NA, Leysens NJ, Zabel BU, Carey JC, Bell GI, Bitoun P, Lindgren C, Stevenson T, Frants RR, van Ommen G, Murray JC (1996) Exclusion of epidermal growth factor and high-resolution physical mapping across the Rieger syndrome locus. Am J Hum Genet 59:1288–1296
Tanizawa Y, Riggs AC, Chiu KC, Janssen RC, Bell DS, Go RP, Roseman JM, Acton RT, Permutt MA (1994) Variability of the pancreatic islet beta cell/liver (GLUT 2) glucose transporter gene in NIDDM patients. Diabetologia 37:420–427
Triggs-Raine BL, Kirkpatrick RD, Kelly SL, Norquay LD, Cattini PA, Yamagata K, Hanley AJ, Zinman B, Harris SB, Barrett PH, Hegele RA (2002) HNF-1alpha G319S, a transactivation-deficient mutant, is associated with altered dynamics of diabetes onset in an Oji-Cree community. Proc Natl Acad Sci USA 99:4614–4619
Voglmaier SM, Bembenek ME, Kaplin AI, Dorman G, Olszewski JD, Prestwich GD, Snyder SH (1996) Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc Natl Acad Sci USA 93:4305–4310
Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P (2000) The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 24:291–295
Acknowledgements
We thank the patients and their family for their participation in this study. We also express our gratitude to Ms. Kazumi Miyazaki, Naoko Takaki, and Yasuko Noguchi for their excellent technical assistance. This work was supported in part by CREST, Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kamimura, J., Wakui, K., Kadowaki, H. et al. The IHPK1 gene is disrupted at the 3p21.31 breakpoint of t(3;9) in a family with type 2 diabetes mellitus. J Hum Genet 49, 360–365 (2004). https://doi.org/10.1007/s10038-004-0158-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-004-0158-z
