
Abstract
Density functional theory calculations of 1H and 13C chemical shifts are used to assign the relative configurations of isohirsut-1-ene, isohirsut-4-ene and tsukubadiene, three terpenes previously isolated from an engineered Streptomyces.
Similar content being viewed by others
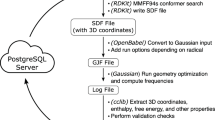
Introduction
The diversity among known terpenes and terpene derivatives (terpenoids) is a testament to the power of terpene synthases in transforming simple biosynthetic building blocks into complex, stereodense natural products.1, 2, 3, 4, 5, 6, 7 Despite decades of work, scientists continue to isolate and identify terpenes and terpenoids with new carbon frameworks from plants, fungi and bacteria.2, 3, 4, 5, 6, 7, 8, 9, 10 Moreover, the capacity of wild-type terpene synthases can be further magnified through genetic engineering. For instance, Cane, Ikeda and co-workers recently described the frameworks of a variety of new terpenes isolated from an engineered Streptomyces host, although relative configurations for many were not fully assigned.11 Here we show how NMR chemical shifts computed with density functional theory (DFT) methods can be used to assign the relative configurations of such structures, using the three structures in Scheme 1 as representative examples.12, 13, 14 This work builds on our past work on confirming, assigning and reassigning the structures of terpenes15, 16, 17 and terpenoids,18, 19 and provides a missing piece of the structural puzzle defined by Cane and Ikeda. This work represents only the second case of assigning the relative stereochemical configuration of complex hydrocarbon natural products, that is, those lacking polar functional groups, using DFT.15
Results and Discussion
Sesquiterpene 8 (isohirsut-1-ene)
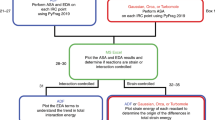
As structure 8 has four stereogenic centers, there are eight possible diastereomers (Scheme 2; each has a corresponding enantiomer, but absolute configuration is not addressed here). First, we constructed and optimized the structures of the eight diastereomers with B3LYP/6-31+G(d,p) in the gas phase (see Methods section). The resulting geometries were then subjected to NMR calculations using SMD(CHCl3)–mPW1PW91/6-311+G(2d,p). Only structure 8F was found to have more than one conformer close enough in energy to contribute to the overall chemical shifts (here there were three); the shifts reported in Table 1 for 8F therefore represent a Boltzmann-weighted average. The mean absolute deviations (MADs) between the experimental and calculated values were determined for each set of carbon and proton chemical shifts for each diastereomer versus the reported experimental values and are shown in Table 1.11 Structure 8D is the best match to the experimental data, based on MAD values for both 1H (0.10 p.p.m.) and 13C (1.60 p.p.m.). Structure 8D also does not contain any large outliers when comparing to the experimental values. To confirm our assignment, DP4 analysis was performed. This statistical analysis, which makes comparisons on an atom-by-atom basis, indicated that 8F is actually the best match (87.4% probability) in terms of 1H shifts (note that 8F also has a small 1H MAD: 0.12 p.p.m.), but 8D is the best match in terms of 13C shifts (99.6%) and when both 1H and 13C shifts are combined in the same analysis (98.8%). Although there is no consensus as to which shifts are generally more diagnostic—1H or 13C—it is clear that 13C shifts are for compound 8 (based on both MAD and DP4 analysis).
Sesquiterpene 9 (isohirsut-4-ene)
Compound 9 is a constitutional isomer of compound 8. As a result, in addition to assigning the relative configuration as we did for 8, we can also confirm that our analysis, simply based on chemical shifts (even in the absence of coupling constants, DEPT data and so on), would differentiate between the hydrocarbon frameworks of 8 and 9. Compound 9 also has four stereogenic centers, again giving rise to eight diastereomers (Scheme 3). Conformational searching yielded three distinct conformers for 9A and 9D after optimization, but for each case, two of these were >3 kcal mol−1 higher in free energy than the lowest-energy conformer, meaning that they would not contribute significantly to the conformational population. Diastereomer 9D is clearly the best match, in terms of 1H or 13C MADs and DP4 probabilities, for the experimental data (Table 2).
Also, as reflected in the data in Table 3, our computed chemical shifts clearly distinguish between the two constitutional isomers 8 and 9. We compared the conformers that were predicted to be the best match for each isomer with the experimental data for the other, that is, computed shifts for 8D were compared with the experimental shifts for 9 and computed shifts for 9D were compared with the experimental shifts for 8. The MADs for both 1H and 13C were much larger for the incorrect isomers (compare values in Table 3 with those in Tables 1 and 2).
Diterpene 11 (tsukubadiene)
We examined compound 11 because it is larger and more flexible than compounds 8 and 9, and it contains an uncommon nine-membered ring, thereby providing a more challenging test of our methods. Compound 11 also contains four stereogenic centers, leading again to eight diastereomers (Scheme 4). Due to its somewhat flexible nine-membered ring, conformational searching gave many possible conformations for each diastereomer (see Supplementary Information for energies of conformers). After optimization with B3LYP/6-31+G(d,p), five conformers were found for 11A that are within 2.1 kcal mol−1 (free energy) of the lowest-energy conformer found (a conformer that is 2 kcal mol−1 higher in energy accounts for ~2% of the population). For 11B, two conformers were found to be within 0.015 kcal mol−1 of the lowest, whereas the third conformer was 6.5 kcal mol−1 higher in energy. Diastereomer 11C had two relevant conformers (spanning, in this case, a 1.4 kcal mol−1 range), 11D had five relevant conformers (spanning, in this case, a 2.3 kcal mol−1 range), 11E had five relevant conformers (spanning, in this case, a 2.4 kcal mol−1 range), 11F had 10 relevant conformers (spanning, in this case, a 1.9 kcal mol−1 range), 11G had four relevant conformers (spanning, in this case, a 2.0 kcal mol−1 range) and 11H had two relevant conformers (spanning, in this case, a 0.5 kcal mol−1 range). Due to the presence of more than one contributing conformer, a Boltzmann-weighted average was used to represent the computed chemical shifts for each diastereomer of compound 11. Listed in Table 4 are the experimental chemical shifts for compound 11 and the differences between computed shifts for each diastereomer and the experimental values. These results indicate that 11A is the best match to the experimental data, with MADs of 2.12 p.p.m. for 13C and 0.09 p.p.m. for 1H and very high DP4 probabilities. Note that diastereomer 11E actually has a lower 13C MAD, but a higher 1H MAD than 11A, but when comparisons are made on a shift-by-shift basis (DP4 analysis), 13C shifts for 11E are a much poorer match for the experimental data than are those for 11A.
Conclusion
We have shown in this report that 1H and 13C chemical shifts computed using quantum chemistry can be used to assign relative configurations of hydrocarbon natural products with high confidence. Specifically, we disclose here the relative configurations of isohirsut-1-ene, isohirsut-4-ene and tsukubadiene, three terpenes previously isolated by Cane, Ikeda and co-workers from an engineered Streptomyces (three-dimensional structures shown in Figure 1).11 Note that if the experimental chiroptical data were available, quantum chemistry could also be used to assign absolute configurations.20 This work builds on our previous assignment of the structure of rhizathalene,15 providing further evidence that this approach is generally useful for terpenes. It is remarkable that such simple methods can be used to distinguish one exceedingly nonpolar molecule from many slightly different nonpolar molecules with high confidence.

Computed three-dimensional structures of the lowest-energy conformers of (top to bottom) isohirsut-1-ene, isohirsut-4-ene and tsukubadiene.
Methods
Quantum chemical calculations were performed with Gaussian09.21 All structural optimizations and frequency calculations (to verify that structures were minima) were performed with B3LYP/6-31+G(d,p) (gas phase).22, 23 1H and 13C chemical shift calculations were performed using mPW1PW91/6-311+G(2d,p)24 and the SMD implicit continuum solvation model25 for chloroform (the solvent used experimentally). Chemical shifts were scaled as described previously13, 26 using scaling factors (slope=−1.0936 for 1H and −1.0533 for 13C; intercept= 31.8018 for 1H and 186.5242 for 13C) available at the cheshirenmr.info website.27 For methyl groups, averages of the computed values of the three hydrogens were used to compare with the experimental data. DP4 statistical analysis28 was performed to assess the probability that our assignments are correct (given the possible alternatives examined, vide infra). For each diastereomer of each compound, a systematic conformational search was first performed using Spartan10.29 For those searches that resulted in only a few conformers, optimizations and NMR calculations were carried out for all of them. For those searches that resulted in many structures, we first calculated the electronic energy of each using B3LYP/6-31G(d) and then, only for the conformers that were within 15 kcal mol−1 of the lowest-energy conformer, we performed optimization with B3LYP/6-31+G(d,p) and NMR calculations (see Supplementary Information for details). To facilitate comparisons with the work of Cane, Ikeda and co-workers, structure numbers correspond to the numbers used in reference 11. Also, note that the chemical shifts for geminal dimethyl groups and diastereotopic CH2 protons are computed separately, but these were averaged to compare with reported values (see Supplementary Information for additional details).

Two sesquiterpenes and a diterpene from reference 11; structure numbers correspond to the numbers used in this reference.

Eight diastereomers of compound 8. Note that, although we do not address herein absolute configuration, the configuration of C9 is shown as identical for all diastereomers to facilitate comparisons.

Eight diastereomers of compound 9. Note that, although we do not address herein absolute configuration, the configuration of C9 is shown as identical for all diastereomers to facilitate comparisons.

Eight diastereomers of compound 11. Note that, although we do not address herein absolute configuration, the configuration of C9 is shown as identical for all diastereomers to facilitate comparisons.
References
Tantillo, D. J. Biosynthesis via carbocations: theoretical studies on terpene formation (review). Nat. Prod. Rep. 28, 1035–1053 (2011).
Cane, D. E. Enzymatic formation of sesquiterpenes. Chem. Rev. 90, 1089–1103 (1990).
Yamada, Y., Cane, D. E. & Ikeda, H. Diversity and analysis of bacterial terpene synthases (review). Methods Enzymol. 515, 123–162 (2012).
Hong, Y. J. & Tantillo, D. J. How cyclobutanes are assembled in nature – insights from quantum chemistry (review). Chem. Soc. Rev. 43, 5042–5050 (2014).
Zerbe, P. & Bohlmann, J. Enzymes for synthetic biology of ambroxide-related diterpenoid fragrance compounds (review). Adv. Biochem. Eng. Biotechnol. 148, 427–447 (2015).
Chen, F., Tholl, D., Bohlmann, J. & Pichersky, E. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom (review). Plant J. 66, 212–229 (2011).
Cane, D. E. Isoprenoids including carotenoids and steroids. Compr. Nat. Prod. Chem. 2, 155–200 (1999).
Blunt, J. W., Copp, B. R., Keyzers, R. A., Munro, M. H. G. & Prinsep, M. R. Marine natural products (review). Nat. Prod. Rep. 33, 382–431 (2016).
Christianson, D. W. Unearthing the roots of terpenome. Curr. Opin. Chem. Biol. 12, 141–150 (2008).
Davis, E. M. & Croteau, R. Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. Top. Cur. Chem. 209, 53–95 (2000).
Yamada, Y. et al. Novel terepenes generated by heterologous expression of bacterial terpene synthase genes in an engineered Streptomyces host. J. Antibiot. 68, 385–394 (2015).
Nguyen, Q. N. N. & Tantillo, D. J. The many roles of quantum chemical predictions in synthetic organic chemistry (a focus review). Chem. Asian J. 9, 674–680 (2014).
Lodewyk, M. W., Siebert, M. R. & Tantillo, D. J. Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 112, 1839–1862 (2012).
Willoughby, P. H., Jansma, M. J. & Hoye, T. R. A Guide to Small-Molecule Structure Assignment through Computation of (1H and 13C) NMR Chemical Shifts. Nat. Protoc. 9, 643–660 (2014).
Vaughan, M. M. et al. Formation of the unusual semivolatile diterpene rhizathalene by the Arabidopsis Class I terpene synthase TPS08 in the root stele is involved in defense against belowground herbivory. Plant Cell 25, 1108–1125 (2013).
Hong, Y. J. & Tantillo, D. J. How many secondary carbocations are involved in the biosynthesis of avermitilol? Org. Lett. 13, 1294–1297 (2011).
Nguyen, Q. N. N. & Tantillo, D. J. Caryolene-forming carbocation rearrangements. Beilstein J. Org. Chem. 9, 323–331 (2013).
Lodewyk, M. W. et al. The correct structure of aquatolide – experimental validation of a theoretically-predicted structural revision. J. Am. Chem. Soc. 134, 18550–18553 (2012).
Quasdorf, K. W., Huters, A. D., Lodewyk, M. W., Tantillo, D. J. & Garg, N. K. Total synthesis of oxidized Welwitindolinones and (-)-N-methylwelwitindolinone C isonitrile. J. Am, Chem. Soc. 134, 1396–1399 (2012).
Stephens, P. J., McCann, D. M., Devlin, F. J. & Smith, A. B. Determination of the absolute configurations of natural products via density functional theory calculations of optical rotation, electronic circular dichroism, and vibrational circular dichroism: the cytotoxic suberosanone, and suberosenol A acetate. J. Nat. Prod. 69, 1055–1064 (2006).
Gaussian 09, Revision D. 01 (Gaussian, Inc.: Wallingford, CT, USA, 2004).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Adamo, C. & Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: the mPW and mPW1PW models. J. Chem. Phys. 108, 664–675 (1998).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Jain, R., Bally, T. & Rablen, P. R. Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem. 74, 4017–4023 (2009).
CHESHIRE. CHEmical SHift REpository with Coupling Constants Added Too. Available at http://cheshirenmr.info. Accessed on 23 March 2016.
Smith, S. G. & Goodman, J. M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability. J. Am. Chem. Soc. 132, 12946–12959 (2010).
Spartan’10 (Wavefunction, Ind.: Irvine, CA, USA (2010).
Acknowledgements
We gratefully acknowledge support from the US National Science Foundation (CHE-1361807 and CHE030089 (XSEDE program)). This work is dedicated to David Cane, an inspiration to mechanistically inclined natural products chemists and natural product-loving mechanistic chemists alike!
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Nguyen, Q., Tantillo, D. Using quantum chemical computations of NMR chemical shifts to assign relative configurations of terpenes from an engineered Streptomyces host. J Antibiot 69, 534–540 (2016). https://doi.org/10.1038/ja.2016.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.51
