
Abstract
The antibiotic strepturidin (1) was isolated from the microorganism Streptomyces albus DSM 40763, and its structure elucidated by spectroscopic methods and chemical degradation studies. The determination of the relative and absolute stereocenters was partially achieved using chiral GC/EI-MS analysis and microderivatization by acetal ring formation and subsequent 2D-NMR analysis of key 1H,1H-NOESY NMR correlations and extraction of 1H,13C coupling constants from 1H,13C-HMBC NMR spectra. Based on these results, a biosynthesis model was proposed.
Similar content being viewed by others
Introduction
The genus Streptomyces (Order Actinomycetales, colloquial ‘Actinomycetes’) is a widely studied genus of the Actinomycete family. Among all Actinomycetes, Streptomycetes play an important role in drug discovery as from this genus 90% of the more than 3000 known antibiotics have been isolated.1 In comparison with other Gram-positive bacteria like Staphylococcus, Streptococcus and Bacillus, the GC-content of the DNA of Streptomyces is with ∼70% comparatively high. Examples of prominent antibiotics produced by these bacteria are the aminoglycoside streptomycin (antituberculosis), the chromopeptide actinomycin (cytostatic) and the polyketide tetracycline (antibacterial).
Here we report on an antibiotic compound produced by Streptomyces albus that exhibits a specific antibacterial activity against the Gram-negative nitrogen-fixing soil bacterium Azotobacter chroococcum.2 This antibacterial activity has been initially discovered in 1956 by Kutzner3 (see also Flaig and Kutzner4), and, as shown later by Böttiger,5 extends to all other members of the family Azotobacteraceae. However, this antibacterial effect is independent from the nitrogen status of the medium and therefore the nitrogenase system of Azotobacter has been excluded as a molecular target.6 The first isolation of the active compound, named strepturidin (1), and its preliminary structure elucidation has already been described by Steinhaus7 in 1993. Initial attempts to characterize the new antibiotic substance with the above-mentioned features by test reactions proved positive for guanidine-groups, sugars and amino sugars. However, test reactions yielded negative results for aromatic amines, organic acids, amino acids and peptides.
This report presents in detail the cultivation of the producing strain, as well as the isolation and structure elucidation of the nucleoside antibiotic strepturidin (1), by means of HPLC-HR-ESI-(+)-Orbitrap-MS, chiral GC/EI-MS, extensive 2D-NMR spectroscopy including partial assignment of the relative stereochemistry and absolute stereochemistry. Based on the investigations on the structure elucidation and determination of the stereochemistry, we postulated a plausible biosynthesis model for (1).
Results
Producing strain and bacterium for antibiotic assay
S. albus has been extensively characterized in the past, notably by Gordon et al.,6 Böttiger5 and Herrmann.8 This streptomycete species can be easily isolated from its natural habitat, that is, self-heated hay or compost, and can be rapidly identified by classical properties. The strain DSM 40763 has been selected from the numerous representatives of this species, which are kept in worldwide culture collections. For the antibacterial assay, the strain A. chroococcum DSM 281 has been employed.
Cultivation and isolation of strepturidin (1)
S. albus DSM 40763 was cultivated as described by Herrmann.8 With a seed culture of 200 ml, a 10 l preculture was inoculated to be used subsequently to inoculate the production fermenter containing 80 l of complex medium (mannitol 10 g l−1, soy meal (full-fat) 10 g l−1, meat extract 5 g l−1, sodium chloride 1 g l−1, calcium carbonate 1 g l−1, tegosipon 10 ml). The cultivation was carried out at 37 °C at 300 r.p.m. and an aeration of 0.5 v.v.m. Growth and antibiotic production were monitored by wet cell mass weight and HPLC analysis, respectively. After 26 h, 130 g l−1 wet cell mass and an antibiotic titer of 92 mg l−1 were determined. The culture broth was harvested by centrifugation, sodium heptanesulfonate was added to the supernatant and the crude antibiotic obtained by ion-pair extraction on Amberlite XAD-16 (Sigma-Aldrich, Taufkirchen, Germany). From the eluate of the resin an excess of sodium heptanesulfonate was extracted as an ion-pair with methyltrioctylammonium chloride using dichloromethane as the solvent. Further purification was carried out by preparative RP18 HPLC and desalting with Amberlite XAD-16 followed by CM-Sephadex NH4+ chromatography as described below. After purification, 1.76 g of pure strepturidin (1) was obtained as a white powder with no defined m.p.
Structure elucidation by means of MS and NMR
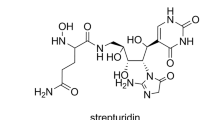
The exact molecular mass of (1) (m/z found 486.19193 [M+H]+) derived from the HR-ESI-(+)-Orbitrap-MS analysis (m/z calcd 486.18227 [M+H]+, Δm 4.883 p.p.m.) gave a molecular formula of C17H27O9N8 [M+H]+. The IR spectrum showed absorptions for amine and hydroxyl groups (3341, 2928 cm−1) and amide carbonyls (1662 cm−1) but no characteristic bands for aromatic hydrocarbons were detected below 900 cm−1 (C-H deformation vibration). The UV spectrum showed a maximum at λ=262 nm (ɛ 3.89) and displayed characteristic absorptions for heterocycles such as nucleobases. Subsequently to the determination of these parameters, the structure elucidation of (1) (Figure 1) was established from extensive use of 1H- and 2D-NMR experiments (1H,1H-COSY, 1H,13C-HMBC, 1H,13C-HSQC, 13C-NMR and 13C-DEPT) in d6-DMSO that revealed a composition of the molecule from four distinct building blocks. The chemical shifts are summarized in Table 1. In the following a more detailed analysis of the NMR data is given.

Structure of strepturidin (1). Bold lines indicate 1H,1H-COSY correlations. Blue arrows indicate 1H,13C-HMBC correlations. Asterisks mark the five stereogenic centers. The list of chemical shifts is summarized in Table 1.
The structure elucidation of (1) commenced with the interpretation of the 1H-NMR (Table 1) and 13C-DEPT NMR spectra.7 The 1H-NMR spectrum of (1) reveals 14 proton NMR signals, with 8 aliphatic methylene (CH2) protons, 5 aliphatic protons and 1 olefinic methine (CH) proton. In analogy to the 1H-NMR, the 13C-DEPT NMR spectrum reveals the corresponding signals of four methylene, six methine and, in addition, seven quaternary carbon signals.7 No primary carbons as represented by methyl groups were detected.
The NMR data of (1) can be formally divided into four spin systems. The first spin system was identified as the glutamine (Gln) Gln-Nα-hydroxamate with four methylene protons at δH-16 2.07 p.p.m. (t, 3JH-16,H-15=7.2 Hz, 2H) and at δH-15 1.9 p.p.m. (m, 2H) and a single aliphatic methine proton at δH-14 4.7 p.p.m. (t, 3JH-14,H-15=3.3 Hz, 3JH-14,NH=8.6 Hz, 1H). The Gln-Nα-hydroxamate spin system is linked through its α-carbonyl at δC-13 169.8 p.p.m. to form an amide (δNH-12 8.4 p.p.m. (b, 1H)) with the amino group of the subsequent spin system identified as a triol-containing carbon chain, an open-chain aldopentose derivative named diamino aldopentose. The linkage was assigned from the methylene protons of the diamino aldopentose alkyl spin system at δH-11 3.2/3.5 p.p.m. (br, 2H; ABX-part, AB-part with broad lines) correlating to δC-13 169.8 p.p.m. in the 1H,13C-HMBC NMR spectrum.7 From the remaining four aliphatic proton signals present in this spin system, three methine protons (CH) at δH-7 4.3 p.p.m. (d, 3JH-7,H-8=5.5 Hz, 1H), δH-9 3.6 p.p.m. (dd, 3J=5 Hz, 1H) and δH-10 3.7 p.p.m. (ddd, 3J=4 Hz, 1H) are bound to three carbons bearing hydroxyl groups. One additional aliphatic methine proton at δH-8 3.8 p.p.m. (dd, 3J=5.4 Hz, 1H) is connecting a third spin system identified as an 2-aminoimidazol-5-one moiety through 1H,13C-HMBC correlations to the quaternary carbons at δC-2′ 157.6 p.p.m. and δC-5′ 168.1 p.p.m., respectively. In addition, the 2-aminoimidazol-5-one spin system accommodates the remaining two methylene protons of structure (1) at δH-4′ 4.2 p.p.m. (s, 2H). The final spin system constitutes the nucleobase uracil that is attached via a C-C-bond to the open-chain diamino aldopentose moiety similar to the C-linkage of the ring ribose to uracil in pseudouridine (ψ). This finding is corroborated by the presence of only one single characteristic olefinic methine proton at δH-6 7.60 p.p.m. (s, 1H) as a part of the uracil structure. These data are complemented by the quaternary carbon signals at δC-2 155.9 p.p.m., δC-4 165.4 p.p.m., δC-5 109.1 p.p.m. and the olefinic methine signal at δC-6 145.4 p.p.m. as part of the nucleobase uracil. The complete structure elucidation is further complemented by 1H,13C-HSQC NMR data7 yielding the assigned protons together with their corresponding carbon atoms and the correlations within the spin parts by the 1H,1H-COSY experiment.7 Figure 1 shows the structure of strepturidin (1) with the key 1H,13C-HMBC contacts with blue arrows and the 1H,1H-COSY correlations in bold lines.
Assignment of the absolute stereochemistry in the Gln-Nα-hydroxamate moiety
The above structure elucidation of (1) revealed the constitutional formula and left the relative and absolute stereochemistry of the five stereocenters at C-7, C-8, C-9, C-10 and C-14 unassigned (Figure 1). Various attempts to crystalize the substance failed, most likely because of the high polarity, structural flexibility and also instability of the molecule (1). Therefore, the subsequent experiments focused on derivatization reactions to assign a maximum number of stereocenters.
Our efforts started with the carbon C-14 that is the stereocenter of the Gln-Nα-hydroxamate. The stereochemistry at C-14 was determined by reduction of the Nα-hydroxyl amine group with 2N HCl/Zn (2 h, room temperature) and transformation into the corresponding amino acid Gln (Figure 2a), generating Gln-strepturidin (2). The reduction was followed by HPLC-HR-ESI-(+)-Orbitrap-MS (Figure 2b) and HPLC-LR-ESI-(+)-MS2 (Supplementary Figure S3). Subsequently, reduced Gln-strepturidin (2) (Figure 2a) was hydrolyzed (6N, HCl, 24 h, 110 °C, under vacuum) to obtain the glutamic acid (Glu) that was derivatized to the N-trifluoroacetyl 2-propyl ester. Amino acid analysis by chiral GC/EI-MS on a LIPODEX E (Macherey-Nagel, Düren, Germany) column (Figure 2c) revealed that the N-trifluoroacetyl Glu 2-propyl ester has (S)-configuration. Based on these results, the stereocenter at C-14 in (1) is also (S)-configured.

(a) Scheme for preparative microreactions performed with strepturidin (1). Formation of monoacetal-strepturidin (3) with 1,1-dimethoxybenzene and camphorsulfonic acid for 2D-NMR experiments. Reduction of strepturidin (1) to Gln-strepturidin (2) for chiral GC/EI-MS analysis. (b) HR-HPLC-ESI-(+)-Orbitrap-MS data of the compounds (1–3). (c) chiral GC-chromatogram on a LIPPDEX E column. Enantiomer analytics of (R/S)-N-trifluoroacetyl Glu 2-propyl esters (Rt-(S)=44.56 min, Rt-(R)=44.11 min) after hydrolysis and derivatization of Gln-strepturidin (2). The EI-MS spectrum of N-trifluoroacetyl (S) Glu 2-propyl ester at Rt=44.56 min is shown in Supplementary Figure S4B. Asterisks mark the stereocenters in strepturidin (1).
Assignment of the relative stereochemistry in the aldopentose moiety
The relative configuration of the remaining four stereocenters in the central part of the molecule C-7 to C-10 could not be derived by the 13C-NMR spectroscopic database approach developed by Kishi and coworkers9 as these databases are only applicable for 1,3-acetals with a methylene group (CH2) connecting the diol groups. In order to reduce the polarity and to increase the rigidity of the molecule for additional NMR spectroscopy experiments of (1), we considered transformation of the 1,3-diol unit into the corresponding 1,3-acetal using 1,1-dimethoxyethylbenzene and camphorsulfonic acid10 (Figure 2a) to give the monoacetal-strepturidin (3) (m/z 589.2 [M+H]+). The monoacetal-strepturidin (3) was isolated by preparative reversed-phase C18-HPLC (Supplementary Figure S1).
The formation of the six-membered 1,3-acetal of compound (3) was proven by 1H-NMR (Supplementary Figure S5) and 2D-NMR experiments (Supplementary Figures S7–S10; 1H,1H-COSY, 1H,13C-HSQC, 1H,13C-HMBC and 1H,1H-NOESY). Application of Mosher’s method for assignment of the absolute configuration determination on the free hydroxyl group at C-10 on the acetal-protected compound (3) has been unsuccessful and no product formation could be detected by HPLC-HR-ESI-(+)-Orbitrap-MS (data not shown).
The formation of the six-membered 1,3-acetal ring present in (3) allowed the analysis of the relative configuration of the triol moiety, using proton–proton coupling constants directly from 1H-NMR data. In addition, selTOCSY-NMR data (Supplementary Figure S9) and long-range couplings from 1H,1H-NOESY data (Figures 3e and f) were used. Because of a lack of strong NOE contacts between the methyl-group protons of δMe-1″ 1.55 p.p.m. and the protons H-6, H-7 and H-9 (Figure 3b and Table 1), an axial position of this group is not plausible and we assume a chair conformation model for the six-membered 1,3-acetal ring with the methyl group in equatorial and the phenyl group in axial position (Figure 3a and Table 1). Furthermore, the small homo-nuclear proton–proton coupling constant for 3JH-7,H-8=3.3 Hz and 3JH-8,H-9=6.3 Hz (48–62° and 30–50° on the basis of the equation of Karplus11 excludes an axial–axial (3Jaa) configuration between the neighboring protons (Figure 3a). The measurements of proton–carbon coupling constants by extraction of 1H,13C-HMBC traces in F2 dimension for the correlations 3JH-8,C-5 and for 3JH-8,C-10 (Figures 3c and d) yield, in both cases, high coupling constants of 3JH,C≈10–13 Hz. Although these measurements are only guidelines, they are in line with above data suggesting an axial orientation of H-8/C-5 and of H-8/C-10, respectively (Figure 3a). Further support for the suggested conformation (Figure 3a) is given by the analysis of 1H,1H-NOESY NMR data (Supplementary Figure S10) with a mixing time of 600 ms. The data showed similarly weak NOE effects from the equatorial methyl protons at δMe-1″ 1.55 p.p.m. to the axial H-8 proton and to the equatorial H-7/H-9 protons (Figure 3f). In addition, a strong NOE effect from the uracil proton δH-6 7.60 p.p.m. to H-7 and a weak NOE to H-8 and H-9 only occur in the case of the postulated arrangement (Figure 3e). Based on these results, we can deduce the relative configuration for the two hydroxyl groups at C-7 and C-9 and the 2-aminoimidazol-5-one group at C-8. Hence, both hydroxyl groups at C-7 and C-9 are syn to each other and anti to the 2-aminoimidazol-5-one substituent at C-8 (Figure 4). For the interpretation of the stereochemistry see the discussion part below.

NMR analysis of monoacetal-strepturidin (3). (a) Plausible model for 1,3 acetal based on NMR analysis. Red arrows indicate 1H,1H-NOESY contacts. Bold lines indicate 3JH-8,C-10 and 3JH-8,C-5 correlations. (b) 1H-NMR region for the three key protons H-7, H-8 and H-9 for compound (3) and expanded 1H-NMR region for H-8. (c) Traces in F2 extracted from 1H,13C-HMBC spectra for C-10 and C-5 (d). The coupling constants 3JH-8,C-10≈10–13 Hz and 3JH-8,C-5≈10–13 Hz suggest an axial orientation. (e) Traces in F2 extracted from 1H,1H-NOESY spectra for H-6. (f) Traces in F2 extracted from 1H,1H-NOESY spectra for the methyl group Me-1″. The weak NOE between H-6 and H-8 in (e) cannot be explained with the conformation shown in (a). Probably, a second conformer with H-8 and uracil in equatorial position exists.

Proposed biosynthesis pathway of strepturidin (1).
Biological activity
Bioactivity testing of pure strepturidin (1) showed the expected activity against A. chroococcum but no activity against the Gram-negative Escherichia coli and the Gram-positive Staphylococcus aureus or Streptomyces glaucescens.6 In an agar diffusion assay on A. chroococcum plates, 40 μg/paper disc produced a 13 mm inhibition zone. Cytotoxicity against the mouse fibroblast cell line L929 was >40 μg ml−1, indicating very low toxicity.
Discussion
With regard to its chemical structure, strepturidin (1) belongs to the group of nucleoside antibiotics. The latter commonly target bacterial peptidoglycan cell wall biosynthesis and fungal chitin wall biosynthesis.12 The group of nucleoside natural products contain a vast number of representatives including well-known compounds, such as tunicamycin, mureidomycin A, liposidomycin B, pacidamycin and nikkomycin Z.12 The common core unit of such nucleoside antibiotics is a uridine nucleotide consisting of a 1′-N-glycosylation of uridine with a (3′-deoxy)-ribose unit.12 Unlike the above-mentioned nucleoside antibiotics, strepturidin (1) is a C-nucleoside with the C-glycosylation at the 5′ position of uracil linked to a linear diamino aldopentose of which we partially assigned the relative stereochemistry. Based on building blocks available from primary metabolism and biosynthetic analogies to other nucleoside antibiotics, we propose a biosynthesis model for (1) as outlined in Figure 4, of which the sequence of intermediate biosynthetic steps may differ: the biosynthetic logic suggests uridine monophosphate (I) with ribose as precursor of the aldopentose-moiety of strepturidin (1). This is in line with the NMR data (Figure 3) that suggest a syn-configuration of C-7 and C-9 and an anti-configuration of C-8 in (1). Therefore, from the four aldopentoses, arabinose and xylose are excluded as likely precursors. Considering that lyxose is a very rare aldopentose in nature,13 this strongly favors ribose as the most likely precursor. As a subsequent step, isomerization of uridine monophosphate (UMP) (I) to pseudouridine (ψ-MP) (II) occurs that is catalyzed by a ψ synthase-type enzyme.14 Such a biosynthetic step has been described for one of the first reported C-nucleosides, 5-β-D-ribofuranosyluracil known as pseudouridine (ψ). Interestingly, C-nucleosides are known to inhibit important enzyme processes, as is shown by the recent discovery of the first C-nucleoside hepatitits C virus polymerase inhibitor GS-6620.15 Further important C-nucleosides are ezomycin B2 (antifungal),16 showdomycin (antibiotic, cytotoxic),17 formycin (antitumor antibiotic)18 and pyrazofurin (antiviral).19 In this context, it is worth mentioning that (1) shows antibiotic activity exclusively against A. chroococcum. In subsequent steps, a nucleotidase could catalyze the hydrolysis of ψ-MP (II) to ψ (III), which finds its analogy in the biosynthesis of caprazamycin and tunicamycin:12 The proceeding oxidation of the terminal 5′OH-group to the 5′aldehyde is followed by a transamination to ψ-5′-amino (IV). Subsequently, ring opening at C-2 by formal addition of H2O occurs while the stereocenter at C-10 is maintained.20 The attachment of the unusual 2-aminoimidazol-5-one building block in (VI) could be derived from condensation of glycine (Gly) and carbamoyl phosphate to the 2′-position of the 2′,5′-diamino aldopentose in (V). Therefore, we hypothesize an enzyme with a transferase-like action to form the 2′-amino group in the aldopentose of (IV) to the intermediate ψ-2′,5′-diamino (V), in analogy to the described 3′-amino formation in the proposed biosynthetic pathway of puromycin.21 Interestingly, the 2-amino pentose formation in Streptomyces alboniger by enzymatic amination is known and described to occur in cis-configuration to the 3′-hydroxyl group that may also define its stereochemistry.22 Also, a similar derivative of 2-aminoimidazol-5-one, the 4-formyl-4-imidazolin-2-one, is found in the nikkomycin biosynthesis, derived from histidine.12 Finally, the amino acid Gln, the precursor of the Gln-Nα-hydroxamate, which forms a peptide bond between its carboxylic group and the primary 5′amine group of the aldopentose building block in (VI). This is assumed to be performed by a non-ribosomal peptide synthetase (NRPS) to form (VII) before or after constituting the Nα-hydroxamate through hydroxylation with an putative amino-monoxoygenase to strepturidin (1). Interestingly, Nα-hydroxamate is rare in nature, whereas most hydroxamate group modifications were found in the side chain of amino acids.23, 24 Although hydroxamates in general display siderophore-like functions,25 common also among other natural products like catecholate and carboxylate siderophores, the role of this moiety in strepturidin is still elusive.
In summary, the structure elucidation of strepturidin (1) renders an unprecedented structure of a novel nucleoside-type antibiotic with an exclusive antibacterial activity against A. chroococcum. Although the biosynthesis of strepturidin is still unknown, we propose similarities to biosynthesis pathways of other (C)-nucleoside antibiotics as mentioned above.
Methods/Experimental section
Producing strain
Large-scale cultivation of strain S. albus (DSM 40763) was performed by Steinhaus7 in collaboration with the Bio-Plant of the GBF (Gesellschaft für Biotechnologisches Forschung) in 1993. A preculture was prepared in a 15 l Giovanola bioreactor (Giovanola Freres, Monthey, Switzerland) charged with a complex medium consisting of mannitol 10 g l−1, biopepton 5 g l−1, yeast extract 5 g l−1, meat extract 5 g l−1, calcium chloride 0.7 g l−1 (pH 7.2) and tegosipon 5 ml, and inoculated with 200 ml of a seed culture. After 14 h, only 5 l of this preculture was left because of extensive foaming that, however, was sufficient for subsequent inoculation. For the production of strepturidin (1), 80 l of a complex medium consisting of mannitol 10 g l−1, soy meal (full-fat) 10 g l−1, meat extract 5 g l−1, sodium chloride 1 g l−1, calcium carbonate 1 g l−1 and tegosipon 10 ml in a 100-l bioreactor (MBR, Zurich, Switzerland) was inoculated with the remaining 5 l of the preculture. The cultivation was carried out at 37 °C at 300 r.p.m. and an aeration of 0.5 v.v.m. (volume per volume per min). After 26 h, 130 g l−1 wet cell mass and an antibiotic titer of 92 mg l−1 were determined. The culture broth was harvested by centrifugation, 180 g of sodium heptanesulfonate was added to the 72 l of supernatant, and the solution passed over a column (10 cm diameter) with 4 l of Amberlite XAD-16 at a constant flow of 8 l h−1. Strepturidin (1) was bound as a heptanesulfonate ion-pair, and, after rinsing the column with water (4 l), (1) eluted with MeOH/H2O, 1:1 (v/v). The eluate was concentrated in vacuo, and the remaining aqueous phase was lyophilized to give 106.4 g of crude extract containing 5.3 g of (1) (yield: 80% by HPLC). To reduce the sodium heptanesulfonate load, ion-pair extraction in two batches was performed. Therefore, each batch was dissolved in 200 ml H2O with methyltrioctylammonium chloride (76.6 g) and each batch was extracted twice with 200 ml CH2Cl2. The aqueous phase was lyophilized to give 28.44 g crude extract with 2.36 g of (1) (yield 95%). Preparative HPLC of this material was performed on a Eurosil C-18 column (10 μm, 250 × 50 mm, Knauer GmbH, Berlin, Germany) with a MeOH/67 mM phosphate buffer (pH 5.0) (solvent A) and 10 mM sodium heptanesulfonate (solvent B) gradient starting with 4:96 (solvent A/B), and ending after 5 min with 9:91 (solvent A/B) at a flow of 100 ml min−1. The detection was carried out at the UV detection wavelength λ=260 nm. For each preparative separation, 3.0–3.5 g of the crude extract was dissolved in 20 ml of H2O to obtain in total 110 g of salts, which after lyophilization of the combined peak fractions contained 2.13 g of (1) (yield 90%). Desalting was performed with 15–20 g portions dissolved in 350 ml H2O and loaded on an Amberlite XAD-16 column (bed volume of 370 ml). The column was rinsed with two to three bed volumes of H2O, and (1) eluted with MeOH/H2O, 1:1 (v:v). From the combined fractions, MeOH was evaporated in vacuo to obtain 16.5 g salts containing 1.96 g (1) (yield 92%). Final desalting was achieved by CM-Sephadex A-25 NH4+ column chromatography (amount of gel 72 g, column size 27.2 × 5 cm, flow 9–10 ml min−1, detection at UV detection wavelength λ=254 nm). The two batches (8 g per batch) were dissolved in 250 ml water and applied to the column followed by two to three bed volumes of water with a flow of 9–10 ml min−1. The pure compound (1) eluted with 50 mM NH3 solution as a sharp peak in the chromatogram. The combined fractions were lyophilized to give 1.76 g of pure (1) as white powder (overall yield 27%).
Analytical HPLC of (1) was performed on a Nucleosil RP-18 (7 μm, 250 × 4 mm, Macherey-Nagel) column with the mobile phase acetonitrile/67 mM phosphate buffer pH 5 with 10 mM sodium heptanesulfonate with a flow of 1.4 ml min−1. Detection was carried out at the UV wavelength λ=262 nm and the retention time of (1) at 11.7 min.
MS
HPLC-HR-ESI-(+)-Orbitrap-MS mass spectra were recorded using a LTQ-Orbitrap XL (Thermo Scientific, Bremen, Germany) coupled to an Agilent 1260 HPLC system (Agilent Technologies, Waldbronn, Germany). The HPLC system was equipped with a Hypersil-Gold column (5 μm, 50 × 2.1 mm, Thermo Scientific, Bremen, Germany). A H2O/MeOH (solvent A/solvent B)+0.1% HCOOH gradient was used starting with 5% B and increasing to 100% B in 6 min, holding 100% B for 4 min, flushing to starting conditions 5% B for 3 min and a flow rate of 0.25 ml min−1.
HPLC-LR-ESI-(+)-MS2 spectra were measured on a QQQ-MS-6460 mass spectrometer (Agilent Technologies) coupled to an Agilent 1290 UHPLC-system (Agilent Technologies). The HPLC system was equipped with an Eclipse Plus C18 RRHD column (1.8 μm, 2.1 × 50 mm, Agilent Technologies). A H2O/ACN (solvent A/solvent B)+0.1% HCOOH linear gradient was used starting with 5% B and increasing to 100% B in 6 min, holding 100% B for 4 min, flushing to starting conditions 5% B for 3 min and a constant flow rate of 0.25 ml min−1.
NMR spectroscopy
The 1D- and 2D-NMR spectra were recorded on a Bruker Avance III 500 spectrometer (Bruker, Karlsruhe, Germany) equipped with a broadband inverse detection probe with Z-Gradient at 500 and 125 MHz for 1H and 13C, respectively. Sample was dissolved in 600 μl d6-DMSO for NMR experiments. Chemical shifts are given relative to TMS. For the calibration, the residual solvent peaks of d6-DMSO (2.50 p.p.m. (1H) and 39.5 p.p.m. (13C)) were used. All experiments were performed at T=298 K.
Synthesis of Gln-strepturidin (2)
To strepturidin (1) (5 mg, 0.011 mmol) dissolved in 2 M HCl (5 ml), Zn dust (0.1 mg) was added in small portions. The extent of reduction of the Nα-hydroxamate group of (1) was followed by HPLC-MS. After 24 h, the sample was centrifuged. The supernatant was purified by C18 solid-phase extraction), washing with H2O and eluting with 100% MeOH. The compound was concentrated in vacuo and then freeze-dried to obtain a yellow oily material. Compound (2) was used for total hydrolysis, derivatization and subsequent chiral GC/EI-MS analytics described in the section below.
Synthesis and purification of monoacetal-strepturidin (3)
The procedure was mainly adopted from the work published by Jundt et al.10 Strepturidin (1) (36 mg, 0.07 mmol), 1,1-dimethoxyethylbenzene (1.15 ml, 0.7 mmol) and camphorsulfonic acid (1.2 mg, 0.005 mmol) were dissolved in 1 ml of d6-DMSO. After stirring overnight at 50 °C, saturated Na2CO3 solution was added to quench the reaction. The mixture was freeze-dried and the residue was dissolved in MeOH and separated by C18-HPLC to yield monoacetal-strepturidin (3) (5 mg, 0.008 mmol, 11% overall yield). Preparative HPLC was performed on an Agilent HPLC 1100 system (Agilent) equipped with a Grom-Sil-120-ODS column (5 μm, 250 mm × 20 mm). A H2O/MeOH (solvent A/solvent B) gradient with a constant flow rate of 15 ml min−1 was used starting with 20% B and increasing to 80% B in 40 min, holding 100% B for 5 min, flushing to starting conditions 20% B for 3 min. Peak collection was carried out at the UV detection wavelength λ=254 nm. The chromatogram is shown in Supplementary Figure S1.
Total hydrolysis of Gln-strepturidin (2) for chiral GC/EI-MS analysis
Total hydrolysis of Gln-strepturidin (2) (0.1 mg) was performed at 110 °C in aqueous 6 M hydrochloric acid solution (200 μl, grade for amino acid analysis; Sigma-Aldrich) under vacuum for 24 h in glass ampoules. After 24 h, the hydrochloric acid was removed in a gentle stream of nitrogen. Compound (2) was semi-purified by C18 solid-phase extraction before chiral GC/EI-MS analysis.
Chiral GC/EI-MS analysis
Chiral GC/EI-MS analysis from the total hydrolysate of Gln-strepturidin (2) was performed on a GC800 Top Voyager (Thermo Finnigan, Waltham, MA, USA) GC-mass spectrometer equipped with a chiral LIPODEX E column (25 m, I.D. 0.25 mm). The scan range was 45–465 amu, detection temperature 210 °C, source temperature 220 °C, ionization energy 70 eV and injection temperature 250 °C. Helium was used as a carrier gas with constant pressure at 60 kPa. Temperature program: 70 °C (2 min isotherm), 90 °C, 3 °C per min (15 min isotherm), 140 °C, 3 °C per min (10 min isotherm), 200 °C, 3 °C per min (1 min isotherm). The injection volume was 1 μl.
Antimicrobial assay
Agar test plates inoculated with A. chroococcum were prepared according to Reis.26 Samples to be tested were applied as 20 μl solutions on paper discs of 6 mm diameter. After incubation for 20 h at 30 °C, the diameter of the inhibition zones was measured. The minimum inhibitory amount of pure strepturidin was 1.7 μg, giving an 8 mm inhibition zone.
Cytotoxicity assay
The inhibitory activity of strepturidin (1) was tested with mouse fibroblasts L-929 (DSMZ ACC 2). Next, 60 μl of a serial dilution of the compound were added to 120 μl aliquots of a cell suspension (50 000 cells per ml) in 96-well microplates. The plates were incubated at 37 °C and 10% CO2 for 6 days. After that time, the MIC was estimated visually using an inverted microscope.
Supporting information
MS data and 2D NMR data are free of charge available via Internet at http://pubs.acs.org.
References
Watve, M. G., Tickoo, R., Jog, M. M. & Bhole, B. D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 176, 386–390 (2001).
Becking, J. H. in The Prokaryotes eds Balows A., Trüper H. G., Dworkin M., Schleifer K. H., 3144–3170 (1991).
Kutzner, H. J. Beitrag zur Systematik und Ökologie der Gattung Streptomyces Waksman & Henrici. PhD thesis, Landwirtschaftliche Hochschule, Hohenheim, Germany (1956).
Flaig, W. & Kutzner, H. J. Beitrag zur Systematik der Gattung Streptomyces Waksman & Henrici. Arch. Microbiol. 35, 105–138 (1960).
Böttiger, V. Untersuchungen an Streptomyces albus und seinem Antibiotikum. PhD thesis, Technische Universität Darmstadt, Germany (1982).
Gordon, R. E., Gray, T. R. E. & Parkinson, B. in. The taxonomy of soil bacteria. The Ecology of Soil Bacteria (eds Gordon, R. E. et al.) 293–321 (Liverpool University Press: Liverpool, United Kingdom, 1967).
Steinhaus, B. Isolierung und Strukturaufklärung von Strepturidin - ein neues Antibiotikum aus Streptomyces albus. PhD thesis, Technische Universität Braunschweig, Germany (1993).
Herrmann, D. Zwei neue Antibiotika aus Streptomyces albus – Untersuchungen zur Optimierung der Biosynthese und Stammverbesserung. PhD thesis, Technische Universität Darmstadt, Germany (1991).
Lee, J., Kobayashi, Y., Tezuka, K. & Kishi, Y. Toward creation of a universal NMR database for the stereochemical assignment of acyclic compounds: proof of concept. Org. Lett. 13, 2181–2184 (1999).
Jundt, L. et al. Isolation and structure elucidation of cruentarens A and B - novel members of the benzolactone class of ATPase inhibitors from the myxobacterium Byssovorax cruenta. Eur. J. Org. Chem. 22, 5036–5044 (2006).
Karplus, M. Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc. 85, 2870–2871 (1963).
Winn, M., Goss, R. J., Kimura, K. & Bugg, T. D. Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 27, 279–304 (2010).
Khoo, K. H. et al. Chemistry of the lyxose-containing mycobacteriophage receptors of Mycobacterium phlei/Mycobacterium smegmatis. Biochemistry 35, 11812–11819 (1996).
Cortese, R., Kammen, H. O., Spengler, S. J. & Ames, B. N. Biosynthesis of pseudouridine in transfer ribonucleic acid. J. Biol. Chem. 25, 1103–1108 (1974).
Cho, A. et al. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J. Med. Chem. (e-pub ahead of print 1 May 2013; doi:10.1021/jm400201a).
Hanessian, S., Dixit, D. M. & Liak, T. J. Studies directed toward the total synthesis of the ezomycins, the octosyl acids and related antibiotics. Pure Appl. Chem. 53, 129–148 (1981).
Darnall, K. R., Townsend, L. B. & Robins, R. K. The structure of showdomycin, a novel carbon-linked nucleoside antibiotic related to uridine. Proc. Natl Acad. Sci. USA 57, 548–553 (1967).
Ochi, K., Yashima, S. & Eguchi, Y. Biosynthesis of formycin. Incorporation and distribution of 13C-, 14C-, and 15N-labeled compounds into formycin. J. Biol. Chem. 254, 8819–8824 (1979).
Westhead, J. E. & Price, H. D. Quantitative assay of pyrazofurin a new antiviral, antitumor antibiotic. Antimicrob. Agents Chemother. 5, 90–91 (1974).
Prior, J. J. & Santi, D. V. On the mechanism of the acid-catalyzed hydrolysis of uridine to uracil. Evidence for 6-hydroxy-5,6-dihydrouridine intermediates. J. Biol. Chem. 259, 2429–2434 (1984).
Tercero, J. A., Espinosa, J. C., Lacalle, R. A. & Jiménez, A. The biosynthetic pathway of the aminonucleoside antibiotic puromycin, as deduced from the molecular analysis of the pur cluster of Streptomyces alboniger. J. Biol. Chem. 3, 1579–1590 (1996).
Rebello, P. F., Pogell, B. M. & Mukherjee, P. P. Formation of 2-amino-2-deoxy-d-ribose 5-phosphate and 2-amino-2-deoxy-d-lyxose 5-phosphate by extracts of Streptomyces alboniger. Biochim. Biophys. Acta 177, 468–485 (1969).
Barona-Gómez, F., Wong, U., Giannakopulos, A. E., Derrick, P. J. & Challis, G. L. Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J. Am. Chem. Soc. 126, 16282–16283 (2004).
Agnoli, K., Lowe, C. A., Farmer, K. L., Husnain, S. I. & Thomas, M. S. The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function sigma factor which is a part of the Fur regulon. J. Bacteriol. 188, 3631–3644 (2006).
Neilands, J. B. Siderphores: structure and function of microbial iron transport compounds. J. Biol. Chem. 270, 26723–26726 (1995).
Reis, K. H. Versuche zur Aufreinigung eines von Streptomyces albus gebildeten Antibiotikums. Diploma thesis, Technische Universität Darmstadt, Germany (1988).
Acknowledgements
We thank the members of the GBF BioPlant for the cultivation of Streptomyces albus, H Steinmetz for his support with large-scale chromatography, B. Elxnat and Ch Heuer for preparing test plates and Dr F Sasse for cytotoxicity assays. We further thank Dr D Herrmann and T Reinhardt and H-H Reis (TU Darmstadt) for valuable information. Financial support from the Cluster of Excellence (‘Unifying concepts in catalysis’ UniCat) granted by the German Research Council (DFG) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Pesic, A., Steinhaus, B., Kemper, S. et al. Isolation and structure elucidation of the nucleoside antibiotic strepturidin from Streptomyces albus DSM 40763. J Antibiot 67, 471–477 (2014). https://doi.org/10.1038/ja.2014.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.16
Keywords
This article is cited by
-
Characterization of C-nucleoside Antimicrobials from Streptomyces albus DSM 40763: Strepturidin is Pseudouridimycin
Scientific Reports (2019)
-
Discovery, properties, and biosynthesis of pseudouridimycin, an antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase
Journal of Industrial Microbiology and Biotechnology (2019)
-
Nature’s combinatorial biosynthesis and recently engineered production of nucleoside antibiotics in Streptomyces
World Journal of Microbiology and Biotechnology (2017)
-
Endophytic bacteria: a new source of bioactive compounds
3 Biotech (2017)
-
Better visualization and photodocumentation of zone of inhibition by staining cells and background agar differently
The Journal of Antibiotics (2015)
