
Abstract
Streptolydigin is a tetramic acid antibiotic produced by Streptomyces lydicus NRRL 2433 and involving a hybrid polyketide synthase (PKS)–nonribosomal peptide synthetase (NRPS) system in its biosynthesis. The streptolydigin amino-acid precursor, 3-methylaspartate, has been proposed to be condensed to the polyketide portion of the molecule by a NRPS composed by three enzymes (SlgN1, SlgN2 and SlgL). On the other hand, biosynthesis of the polyketide moiety involves the participation of cytochrome P450 SlgO2 for the correct cyclization of the characteristic bicyclic ketal. Independent disruption of slgN1, slgN2, slgL or slgO2 resulted in S. lydicus mutants unable to produce the antibiotic thus confirming the involvement of these genes in the biosynthesis of the antibiotic. These mutants did not accumulate any streptolydigin biosynthesis intermediate or shunt product derived from early polyketides released from the PKS. However, they produced three novel compounds identified as 4-(2-carboxy-propylamino)-3-chloro-benzoic acid, 4-(2-carboxy-propylamino)-3-hydroxy-benzoic acid and 4-(2-carboxy-propylamino)-benzoic acid, which were designated as christolane A, christolane B and christolane C, respectively. These compounds have been shown to exert some antibiotic activity.
Similar content being viewed by others
Introduction
Polyketides and nonribosomal peptides constitute two important groups of secondary metabolites highly relevant as therapeutics for clinical use as many of them are potent antibiotic, antifungal, antitumor, immunosuppressant, antiviral or antiparasitic agents.1, 2, 3, 4, 5 Polyketides are produced by polyketide synthases (PKSs) by step-wise decarboxylative Claisen-type condensation of acyl-CoA precursors that are further modified by different activities such as ketoreductase, dehydratase and enoylreductase in the case of type I PKSs.6, 7, 8 On the other hand, nonribosomal peptides are synthesized by nonribosomal peptide synthetases (NRPSs) using as monomeric building blocks proteinogenic and non-proteinogenic amino acids and other carboxylic acids. NRPSs follow the same chemical logic as PKSs for chain elongation and then the incorporated amino-acid monomers can be modified by epimerization, methyltransferase, reductase or oxidase activities.6, 7, 8, 9 Another group of secondary metabolites, mixed polyketide-nonribosomal peptides, are synthesized by the combined use of NRPSs and PKSs.10 An important issue for the biological activity of these groups of natural products is the tailoring modifications incorporated once the polyketide and/or the nonribosomal peptide core has been synthesized. In particular, the introduction of oxygen-containing functionalities is of great importance as it can provide a base for additional modifications such as methyl, amino or glycosyl transfer and cyclizations.11, 12, 13, 14, 15
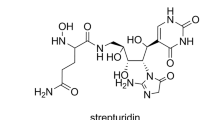
Streptolydigin (Figure 1) is a polyketide-nonribosomal peptide compound member of the tetramic acids family that possesses antibiotic activity.16, 17 The streptolydigin biosynthesis gene cluster was isolated from Streptomyces lydicus NRRL 2433 and the biosynthesis and tailoring modification of 3-methylaspartate, substrate of the streptolydigin NRPS system, have been investigated.18, 19, 20 Three genes were proposed to encode the streptolydigin NRPS: slgN1, slgN2 and slgL. In addition, two genes, slgO1 and slgO2, were found to encode cytochrome P450 enzymes proposed to participate in the generation of streptolydigin bicyclic ketal and epoxide moieties.18

Streptolydigin biosynthesis pathway. A, adenylation domain; A’, adenylation domain SlgN1; ATa, acyl transferase specific for malonyl-CoA; ACP, acyl carrier protein; ATp, acyl transferase specific for methylmalonyl-CoA; C, condensation domain; DH, dehydratase; L, SlgL; M, PKS module; PCP, peptidyl carrier protein; KR, ketoreductase; KR*, inactive ketoreductase; KS, ketosynthase.
In this work, we investigated the involvement of NRPS (slgN1, slgN2 and slgL) and the specific role of cytochrome P450 slgO2 in the biosynthesis of streptolydigin. In addition, three novel compounds, named christolanes, were isolated and characterized from streptolydigin non-producer mutants generated during the development of this work.
Materials and methods
Strains and culture conditions
Bacterial strains used in this work were S. lydicus NRRL 2433, streptolydigin producer, Escherichia coli DH10B (Invitrogen, Fisher Scientific, Madrid, Spain) and ET12567 (pUB307)21 used for subcloning. The growth medium for S. lydicus and mutants was tryptone soya broth. For S. lydicus sporulation and streptolydigin production MA and R5A medium were used, respectively.22 E. coli media were those described in the literature.23 Conjugation of Streptomyces mycelium was performed following the standard procedures.21 When plasmid-containing clones were grown, the medium was supplemented with the appropriate antibiotics: 100 μg ml−1 ampicillin, 20 μg ml−1 tobramycin, 25 μg ml−1 apramycin, 50 μg ml−1 thiostrepton, 10 μg ml−1 tetracycline, 25 μg ml−1 chloramphenicol or 50 μg ml−1 nalidixic acid.
DNA manipulation and plasmids
DNA manipulations were performed according to the standard procedures for E. coli22 and Streptomyces.21 PlatinumPfx DNA polymerase (Invitrogen) and 2.5% dimethylsulphoxide (DMSO) were used for all PCR amplifications. PCR conditions used were 97 °C, 5 min; 30 cycles of 95 °C, 30 sec, 55 °C, 45 sec, and 68 °C, 1 min and a final extension cycle at 68 oC, 10 min. All the PCR products were cloned into pCR-BLUNT (Invitrogen) and then sequenced. Other plasmids used in this work were pUC18 (Invitrogen) and pSL1180 (Amersham Pharmacia Biotech, Europe GmbH, Barcelona, Spain) for routine cloning, pOJ26024 and pOJ260P25 for gene disruption and pEM4T26 for gene expression.
Construction of plasmids for gene inactivation and complementation of mutants
Inactivation of slgN1 was accomplished by amplification of a slgN1 internal fragment of 998 bp by PCR from cosmid Slg4A818 using oligoprimers CRIS7 (5′-AGAATTCAGATCGCCGGTGCGGTAC-3′, EcoRI underlined) and CRIS8 (5′-AAAAAGCTTGGTCGGCGTCCATCTGG-3′, HindIII underlined). The PCR product was cloned into pCR-BLUNT and then sequenced. The resultant plasmid was digested HindIII–EcoRI and the 998-bp fragment cloned into pOJ260 to obtain plasmid pOJN1 used for generation of S. lydicus strain SLMN1.
Plasmid pOJ260P was used to generate different constructs for the inactivation of slgN2, slgL and slgO2. This plasmid places the genes downstream of the insertion point in the mutant strain under the control of the promoter ermE* to avoid possible polar effects. Oligoprimers CRIS27 (5′-AATCTAGACTGGAGACGGACTGCGGC-3′, XbaI underlined) and CRIS28 (5′-AGAATTCACGAGGCACAGCTCGCCG-3′, EcoRI underlined were used for amplifying an internal 829-bp region of slgN2 from cosmid Slg6E5.18 The same cosmid was used for the amplification of a 621-bp slgL internal fragment using oligoprimers CRIS25 (5′-AATCTAGATGCTGGCCGTCGACTACC-3′, XbaI underlined) and CRIS26 (5′-AGAATTCAGTCCCGTGAGCTGACGG-3′, EcoRI underlined). A slgO2 internal fragment of 872 bp was amplified from cosmid Slg4A8 using oligoprimers CRIS11 (5′-AATCTAGACTAGTCGGCTGAGCACCGACCTC-3′, XbaI underlined) and CRIS12 (5′-AGAATTCGTAGTGGATGCCGTGGCC-3′, EcoRI underlined). Finally, each fragment was digested with XbaI–EcoRI and subcloned into the same sites of pOJ260P, downstream of the promoter ermE*. This led to plasmids pOJEN2, pOJEL and pOJEO2 that were used for the generation of S. lydicus mutants SLMN2, SLML and SLMO2, respectively.
The S. lydicus mutants SLMN1, SLMN2, SLML and SLMO2 affected in slgN1, slgN2, slgL and slgO2, respectively, were complemented using the appropriate genes cloned in plasmid pEM4T under the control of ermE* promoter. A 2-kb KpnI-SstI fragment from cosmid Slg4A8-containing slgN1 was cloned in pUC18, then digested EcoRI–BamHI and subcloned in pSL1180 using the same restriction sites. From the last construct, pSLslgN1, an EcoRI-MfeI fragment was obtained and subcloned, in the correct orientation, in pEM4T digested with EcoRI leading to pEM4TslgN1. Oligoprimers CCL5B (5′-AAGGATCCGCTGACCCACCCCGCACC-3′, BamHI underlined) and CCL5EI (5′-AAGAATTCCGCCCCGTTGCCGTTCAG-3′, EcoRI underlined) were used to amplify slgN2 from cosmid Slg6E5. The PCR fragment was cloned in pCR-BLUNT and sequenced leading to pCCL5BEI. All constructs analyzed were found to carry a deletion of the CCL5EI region, while the CCL5B region was correct. To avoid this problem a 1.3-kb BamHI-EcoNI fragment from pCCL5BEI and a 1.3-kb EcoNI-SacI fragment from cosmid Slg6E5 were subcloned together in pUC18 digested with BamHI-SacI. From the former construct, pUCslgN2, a 2.6-kb BamHI–EcoRI fragment was subcloned in pEM4T leading to pEM4TslgN2. Oligoprimers CCL6Bg (5′-AAAAGATCTCGTGAATACATCCCTGAA-3′, BglII underlined) and CCL6EI (5′-AAGAATTCCTTGCTGTGTGTGACGTG-3′, EcoRI underlined) were used to amplify slgL from cosmid Slg6E5. The resultant PCR product was cloned in pCR-BLUNT, sequenced and then subcloned as a BglII–EcoRI fragment in pEM4T digested with BamHI–EcoRI that led to pEM4TslgN2. Oligoprimers CRIS13 and CRIS1418 were used for PCR amplification of slgO2 from cosmid Slg4A8. The resultant PCR product was cloned in pCR-BLUNT, sequenced and then subcloned in BamHI–EcoRI digested pEM4T leading to pEM4TslgO2.
Generation of S. lydicus mutant strains
Constructs pOJN1, pOJEN2, pOJEL and pOJEO2 were introduced into S. lydicus by intergeneric conjugation from E. coli ET12567 (pUB307). Transconjugant S. lydicus strains SLMN1, SLMN2, SLML and SLMO2 were selected for resistance to apramycin and integration in the S. lydicus chromosome was verified by Southern hybridization. Plasmids pEM4T, pEM4TslgN1, pEM4TslgN2, pEM4TslgL and pEM4TslgO2 were introduced in S. lydicus mutants SLMN1, SLMN2, SLML and SLMO2 by intergeneric conjugation and transconjugants were selected for resistance to thiostrepton.
Ultra performance LC (UPLC), LC-MS and isolation of christolanes
Streptolydigin production by S. lydicus wild-type or mutant strains grown on R5A medium was analyzed by UPLC (acquity UPLC equipment with a BEH C18 Waters column of 2.1 × 100 mm) and LC-MS (Alliance chromatographic system coupled to a ZQ4000 mass spectrometer and a Symmetry C18 Waters column of 2.1 × 150 mm) using previously described procedures.18 Detection and spectral characterization of peaks were performed in both cases by photodiode array detection and Empower software (Waters Cromatografia SA, Cerdañola del Vallés, Spain), extracting bidimensional chromatograms at 360 and 300 nm. For structural elucidation of christolanes B (1), C (2) and A (3), they were purified from solid R5A medium agar plates. Liquid cultures of each strain were grown as a seed culture in tryptone soya broth as described above before18 and used to inoculate the agar plates. After 7 days at 30 °C, agar media was extracted with ethyl acetate and concentrated by evaporation using a rotavapor. Extracts were dissolved in 4 ml methanol and then chromatographed as described above.
Physicochemical properties
NMR spectra were recorded at 25 °C on a Bruker AVII-600 using a 5-mm TCI cryoprobe and processed using Topspin2.16 software (Bruker GmbH, Karlsruhe, Germany). 13C NMR spectra were acquired with decoupling over a spectral width of 240 p.p.m. with 64 K data points collecting 6000 accumulations. A 4-s relaxation delay was incorporated between each scan. COSY, TOCSY, HMBC and HSQC were all collected in the phase-sensitive mode. For each of these experiments, 256–512 t1 increments were used. In all, 8 transients were collected for COSY and TOCSY and 48 transients for HMBC and HSQC. The relaxation delays were set to 1.5 s. TOCSY spectra were recorded using the decoupling in the presence of scalar interaction pulse sequence with mixing times (spin-lock) of 80 ms. In HSQC and HMBC experiments, direct JCH couplings were set to 145 Hz, while 6 Hz were used for long range JCH. HMBC spectra were acquired with a low pass J-filter to suppress one-bond correlations.
4-(2-Carboxy-propylamino)-3-hydroxy-benzoic acid, christolane B (1): yellow amorphous powder; 1H NMR (600 MHz, [D6]DMSO, 25 °C): δ=7.33 (dd, 3J (H,H)=8.3 Hz, 4J (H,H)=1.9 Hz, 1 H; CH), 7.24 (d, 4J (H,H)=1.9 Hz, 1 H; CH), 6.55 (d, 3J (H,H)=8.4 Hz, 1 H; CH), 3.63 (dd, 3J (H,H)=7.4 Hz, 3J (H,H)=7.0 Hz, 1 H; CH2), 3.18 (dd, 3J (H,H)=7.4 Hz, 3J (H,H)=7.0 Hz, 1 H; CH2), 2.70 (m), 1.11 (d, 3J (H,H)=7.0 Hz, 3 H; CH3). 13C NMR (150 MHz, [D6]DMSO, 25 °C): δ=176,3, 167.6, 143.0, 141.4, 122.9, 117.1, 113.6, 107.9, 45.3, 38.5 and 14.8. MS (100 eV): m/z (%): 240 (90) [M+].
4-(2-Carboxy-propylamino)-benzoic acid, christolane C (2): yellow amorphous powder; 1H NMR (600 MHz, [D6]DMSO, 25 °C): δ=7.67 (d, 3J (H,H)=9.0 Hz, 2 H; CH), 6.60 (d, 3J (H,H)=9.0 Hz, 2 H; CH), 6.60 (d, 3J (H,H)=9.0 Hz, 2 H; CH), 3.36 (dd, 3J (H,H)=7.1 Hz, 3J (H,H)=7.0 Hz, 1 H; CH2), 3.09 (dd, 3J (H,H)=7.1 Hz, 3J (H,H)=7.0 Hz, 1 H; CH2), 2.64 (q, 3J (H,H)=6.9 Hz, 1 H; CH), 1.13 (d, 3J (H,H)=7.0 Hz, 3 H; CH3). 13C NMR (150 MHz, [D6]DMSO, 25 °C): δ=176.1, 167.5, 152.3, 131.1, 117.0, 110.8, 45.4, 38.6 and 15.0. MS (100 eV): m/z (%): 224 (90) [M+].
4-(2-Carboxy-propylamino)-3-chloro-benzoic acid christolane A (3): yellow amorphous powder; 1H NMR (600 MHz, [D6]DMSO, 25 °C): δ=7.75 (d, 4J (H,H)=1.9 Hz, 1 H; CH), 7.72 (dd, 3J (H,H)=8.8 Hz, 4J (H,H)=1.9 Hz, 1 H; CH), 6.82 (d, 3J (H,H)=8.8 Hz, 1 H; CH), 6.08 (m, 1 H; NH), 3.47 (dd, 3J (H,H)=7.0 Hz, 3J (H,H)=6.0 Hz, 1 H; CH2), 3.26 (dd, 3J (H,H)=7.0 Hz, 3J (H,H)=6.0 Hz, 1 H; CH2), 2.73 (q, 3J (H,H)=7.1 Hz, 1 H; CH), 1.21 (d, 3J (H,H)=7.1 Hz, 3 H; CH3). 13C NMR (150 MHz, [D6]DMSO, 25 °C): δ=176.1, 166.4, 147.3, 130.2, 130.0, 118.2, 117.0, 110.1, 45.3, 38.1 and 14.8. MS (matrix-assisted laser desorption/ionisation-time of flight ): m/z (%): 256 (90) [M−].
Bioactivity testing
Antibiotic activity of christolanes was analyzed via antibiotic disc diffusion assay against Streptomyces albus, Streptococcus pyogenes, Staphylococcus aureus, Micrococcus luteus, E. coli, Pseudomonas aeruginosa, Serratia marcenscens and Klebsiella pneumoniae following previously described procedure.18 Antifungal activity was tested against Candida albicans. In all cases, 2 μg of each compound was used. The minimum inhibitory concentration of S. albus was defined by plating the different strains on 25-ml MA agar plates containing different concentrations of christolanes. The plates were incubated for 5 days at 30 °C.
Antitumor activity of christolanes was tested against the following human tumor cell lines: colon adenocarcinoma (HCT-116 and HT29), non-small cell lung cancer (A549), breast adenocarcinoma (MDA-MB-231), acute T-cell leukemia (JURKAT), pro-myelocytic leukemia (HL60) and cervix carcinoma (HeLa). The mouse embryonic fibroblast cell line NIH/3T3 was used as control to evaluate cytotoxicity against non-malignant cells. Quantitative measurement of cell growth and viability at 24, 48 and 72 h was carried out by using a colorimetric type of assay, using the cell proliferation assay and cytotoxicity assay system Cell Counting Kit-8 (CCK-8) following the manufacturer protocol (Dojindo Molecular Technologies, Europe GmbH Munich, Germany).
Results
Inactivation of streptolydigin NRPS genes
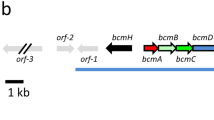
The participation of slgN1, slgN2 and slgL, encoding enzymes with different NRPS domains (Figure 2), in streptolydigin biosynthesis was assessed by their inactivation in S. lydicus (Figure 3). Each of these genes was inactivated by disruption using internal fragments to the genes cloned in plasmids pOJN1, pOJN2 and pOJL, respectively. UPLC analysis at 360 nm of products accumulated by the corresponding mutant strains: SLMN1, SLMN2 and SLML, showed that streptolydigin was not produced in any of them (Figure 3b). Analysis at different wavelengths of the UPLC chromatograms showed the presence of three peaks, in variable amounts, in each of the mutants when the chromatograms were extracted at 300 nm. These new peaks were not observed in S. lydicus wild-type cultures (Figure 3c). The limit of detection of each peak was determined to be of 10 pg. Compounds 1, 2 and 3 with UPLC retention times of 2.81, 3.01 and 3,75 min showed masses of 240, 224 and 258 m/z [M+H]+, respectively, under MS analysis. Production of streptolydigin in these mutants was restored by reintroduction of the corresponding genes under the control of ermE* promoter using plasmids pEM4TslgN1, pEM4TslgN2 and pEM4TslgL, respectively (data not shown).

Sequence aligment of streptolydigin NRPSs and monooxygenase proteins. Alignment of SlgN1 with NRPS proteins containing a discrete adenylation domain: CloN4 from Streptomyces roseochromogenes subsp. oscitans (accession number AAN65233), CouN4 from Streptomyces rishiriensis DSM 40489 (AAG29789) and LnmQ from Streptomyces atroolivaceus S-140 (AAN85530 (a). Alignment of SlgN2 with NRPS proteins: LipNrps from Streptomyces aureofaciens Tü117 (ABB05101), TamD from Streptomyces sp. 307–9 (ADC79642), TrdD from Streptomyces sp. SCSIO1666 (ADY38536) and KirB from Streptomyces collinus Tü365 (CAN89638) (b). Alignment of SlgL with TamC from Streptomyces sp., 307–9 (ADC79641), TrdC from Streptomyces sp., SCSIO1666 (ADY38535), LipX2 from S. aureofaciens Tü117 (ABB05100), KirHI from S. collinus Tü365 (CAN89637) and Orf28 from Streptomyces verticillus ATCC15003 (AAG02344) (c). Alignment of SlgO1 and SlgO2 with cytochomes P450 involved in the biosynthesis of different polyketide compounds showing the sequence around the O2-binding site and the cytochrome P450 cysteine heme-iron ligand signature. TamI from Streptomyces sp., 307–9 (ADC79647), TrdI from Streptomyces sp., SCSIO1666 (ADY38541), OleP from Streptomyces antibioticus (AAA92553) and PimD from Streptomyces natalensis (CAC20932) (d). Aa, amino acids; x, any amino acid

Organization of the streptolydigin biosynthesis gene cluster. Black arrows indicate the three NRPS encoding genes involved in streptolydigin biosynthesis. Gray arrows indicate the two cytochrome P450 encoding genes involved in streptolydigin biosynthesis (a). UPLC analysis at 360 nm of products accumulated by mutants SLMN1, SLMN2, SLML and SLMO2 (b). UPLC analysis at 300 nm of products accumulated by mutants SLMN1, SLMN2, SLML and SLMO2 (c).
Inactivation of slgO2
Cytochrome P450 encoded by slgO2 (Figure 2d) is suspected to participate in the bicyclic ketal formation during streptolydigin biosynthesis. For this reason we attempted the disruption of slgO2 to identify streptolydigin analogs lacking this moiety. Mutant SLMO2 was generated using plasmid pOJEO2. Analysis of SLMO2 culture extracts by UPLC extracting the chromatograms at 360 nm showed no production of streptolydigin or any other putative intermediate (Figure 3b). Furthermore, streptolydigin production in SLMO2 was restored by introduction of plasmid pEM4TslgO2 carrying the corresponding gene under the control of ermE* promoter.
Linear or cyclic intermediates derived from the premature release of products from the streptolydigin PKS or NRPS enzymes were search by UPLC analysis performed at different wavelengths using SLMO2 culture extracts. Only when the wavelength was fixed at 300 nm three peaks with retention times identical to those of compounds 1, 2 and 3, previously identified from mutants SLMN1, SLMN2 and SLML were found (Figure 3c). These peaks were confirmed by MS analysis to have the same masses as reported above. In addition, the production of these compounds was also verified to occur in other S. lydicus mutant strains previously reported: they were detected in streptolydigin non-producers SLM4C1 and SLM96118 and in strains producing small amounts of different streptolydigin analogs: SLM7H13,18 SLMZ,19 SLMM19 and SLME1E2.20 In all cases, compound 3 was consistently the major product accumulated.
Structural characterization of christolanes
Compounds 1, 2 and 3 were purified from cultures of SLMN1 (13, 11 and 38 mg l−1, respectively) and SLMO2 (11, 11 and 50 mg l−1, respectively) and their structural elucidation was carried out using 1D 1H, 1D 13C, 2D 1H COSY, 2D 1H TOCSY, 1H, 13C HSQC edited and HMBC NMR experiments (see Supporting information), The 13C spectra of all three compounds showed two signals at 176 and 167 p.p.m., corresponding to carboxylic acids, the latter being an aromatic one. Furthermore, in the spectra of 1 and 3, six aromatic signals were identified, three of them corresponding to quaternary carbons as could be verified in the HSQC spectrum. This corresponds to a trisubstituted benzene ring and, as one of the substituents is a carboxylic acid, we concluded that the compounds were derivatives of benzoic acid. As could be elucidated from the 2D experiments, the second substituent is 2-carboxy-propylamine, including the second carboxylic acid group. The third substituent in compounds 1 and 3 were a hydroxyl and a chloro, respectively, as we deduced from the masses obtained from MS spectroscopy. In addition, the obtained 1H and 13C chemical shifts fitted very well with the predicted chemical shifts for these compounds (Table 1). In the case of compound 2, we only found four different aromatic carbon resonances, two of them quaternary, from where we deduced that there was a disubstituted symmetric aromatic ring. One of the substituents is the carboxylic acid group while the other is again 2-carboxy-propylamine.
All this information allowed us to identify the compounds as 4-(2-carboxy-propylamino)-3-hydroxy-benzoic acid (1), 4-(2-carboxy-propylamino)-benzoic acid (2) and 4-(2-carboxy-propylamino)-3-chloro-benzoic acid (3), none of them structurally related to streptolydigin (Figure 4a). These novel compounds were named christolane B (1), christolane C (2) and christolane A (3) as, as far as we know, after an extensive structural search using SciFinder (CAS Division of American Chemical Society, Columbus, OH, USA) search tools they have not been previously reported.

Structure of christolanes (a). Antibiotic activity of streptolydigin (S), christolane B (1), C (2) and A (3) against S. albus. Control without antibiotic (C) (b).
Biological activity of christolanes
The three christolanes were tested to verify their biological activity, both antibiotic and cytotoxic. The antibacterial activity was monitored against Gram-positive S. albus, S. pyogenes, S. aureus and M. luteus, Gram-negative E. coli, P. aeruginosa, S. marcenscens and K. pneumoniae and yeast C. albicans using streptolydigin as a control and following the procedure previously described.18 Christolanes B (1), C (2) and A (3) showed a narrow antibiotic activity being only active against S. albus at the concentration tested. The inhibition halos of christolanes with diameters of 1.2, 1.6 and 2 cm, respectively, were smaller than that of streptolydigin, 3.7 cm (Figure 4b). In accordance with the opaque inhibition halos observed in the bioassay, the antibiotic activity of christolanes was found to be very weak being their minimum inhibitory concentrations against S. albus >200 μg ml−1.
The cytotoxicity of christolanes was tested against human tumor cell lines HCT-116, HT29, A549, MDA-MB-231, JURKAT, HL60, HeLa and mouse cell line NIH/3T3. No cytotoxic activity was observed against any of these cell lines at 100 μM.
Discussion
Streptolydigin biosynthesis (Figure 1) has been shown to involve a PKS-NRPS system through the condensation of four units of malonyl-CoA, four units of methylmalonyl-CoA and the incorporation of amino-acid 3-methylaspartate derived from glutamate.18, 19, 20 The streptolydigin NRPS system has been proposed to be formed by SlgN1 and SlgN2. SlgN1 contains a discrete NRPS adenylation (A) domain and similar proteins have been described in leinamycin, clorobiocin and coumermycin A1 pathways.27, 28 SlgN2 is composed by three domains, condensation (C), adenylation (A) and peptidyl carrier protein being the A domain shorter than previously described for other NRPSs.18 It shows high similarity to mono-modular NRPSs involved in the biosynthesis of tetramic acids α-lipomycin29 and tirandamycin.30, 31 In addition, SlgL might be involved in the adenylation process as it has a putative A4 adenylation core motif and shares high similarity with proteins encoded by genes found in α-lipomycin,29 tirandamycin,30, 31 bleomycin32 and kirromycin33 gene clusters. The independent inactivation of streptolydigin NRPS genes slgN1, slgN2 or slgL abrogated the tetramic acid biosynthesis. In addition, no streptolydigin intermediaries or shunt products were detected in culture broths of strains SLMN1, SLMN2 or SLML. These results clearly confirm the involvement of SlgN1, SlgN2 and SlgL in the biosynthesis of streptolydigin, more likely in the incorporation of the amino-acid precursor.
On the other hand, the formation of the bicyclic ketal and it has been proposed to be performed by a cytochrome P450 through oxidation of a C2 hydroxyl group to a ketone in a PKS-bound intermediate followed by the cyclization events.18 Two cytochrome P450s coding genes, slgO1 and slgO2, are located in streptolydigin cluster. SlgO1 and SlgO2 contain the conserved O2-binding site and the C-terminal heme-binding domain characteristic of cytochrome P450s34 In addition, SlgO1 shows a high level of identity with PimD and OleP, monooxygenases shown to participate in the epoxidation of pimaricin and oleandomycin, respectively.34, 35 On the other hand, SlgO1 is more similar than SlgO2 to TrdI and TamI, cytochromes P450 involved in oxidative modifications during the biosynthesis of tetramic acid tirandamycin.30, 31 Disruption of trdI or tamI led to tirandamycin C, an intermediate that lacks any oxidative modifications.30, 31 On the basis of these facts, we propose that SlgO1 might be involved in the introduction of streptolydigin epoxide moiety (Figure 1). According to this, SlgO2 should be in charge of oxidative reactions leading to the formation of streptolydigin bicyclic ketal (Figure 1), point that has been confirmed by inactivation of slgO2. Mutant strain SLMO2 did not produce streptolydigin. Furthermore, no streptolydigin intermediates or derivatives were detected in SLMO2 cultures. If SlgO2 were involved in epoxide formation, we would expected the accumulation of compounds desepoxystreptolydiginone or desepoxystreptolydigin lacking the corresponding oxygenation such as has been observed in other pathways.30, 31, 36
It has been previously described that different linear or cyclic intermediates derived from the premature release of the polyketide chain assembly line could be expected due to the inactivation of genes encoding enzymes involved in cyclization or tailoring modification of the polyketide backbone.37, 38, 39 The presence of such intermediates, or shunt products derived from them, were expected to be produced by strains SLMN1, SLMN2, SLML or SLMO2 affected at two different stages of the streptolydigin biosynthesis but still containing a functional PKS. However, no such compounds were identified. Otherwise, three novel compounds not structurally related to streptolydigin were isolated and characterized: christolanes B (1), C (2) and A (3). They were found to exert antibiotic activity only against S. albus being christolane A (3) the most active one, probably due to the presence of a chlorine moiety at C3. On the other hand, christolane C (2) shows more antibiotic activity than christolane B (1). The unique structural difference between these two compounds is the presence of a hydroxyl moiety at C3 in christolane B.
Christolanes might be produced from a silent or weakly expressed biosynthetic gene cluster as they have not been detected by UPLC analysis of wild-type S. lydicus NRRL 2433. However, after inactivation of streptolydigin biosynthesis genes some common intermediates can be derived for the production of these compounds, being their production clearly detected. This phenomenon has been described in other systems such as the increase of nanchangmycin production by Streptomyces nanchangensis when a different biosynthesis cluster was inactivated,40 or actinorhodin production by Streptomyces coelicolor after decreasing undecylprodigiosin yields.41 A possible christolane pathway could be the condensation of 3-aminoisobutyric and 4-hydroxybenzoic acids or 3-hydroxyisobutyric and 4-aminobenzoic acids through the activity of a phenazine PhzF-like condensing enzyme42 or a C-1027 SgcD5-like amide bond-forming enzyme43 to generate christolane C. This compound might be then hydroxylated to generate christolane B or chlorinated leading to christolane A. The biosynthetic origin of putative christolane precursors might be chorismic acid44, 45, 46 and valine47 or pyridoxyal-catalyzed decarboxylation of 3-methylaspartate,48 issue that would require further experiments to be confirmed.
Accession codes
References
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477 (2007).
Butler, M. S. Natural products to drugs: natural product-derived compounds in clinical trials. Nat. Prod. Rep. 25, 475–516 (2008).
Cragg, G. M., Grothaus, P. G. & Newman, D. J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 109, 3012–3043 (2009).
Olano, C., Méndez, C. & Salas, J. A. Antitumor compounds from actinomycetes: from gene clusters to new derivatives by combinatorial biosynthesis. Nat. Prod. Rep. 26, 628–660 (2009).
Olano, C., Méndez, C. & Salas, J. A. Antitumor compounds from marine actinomycetes. Mar. Drugs 7, 210–248 (2009).
Staunton, J. & Weissman, K. J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18, 380–416 (2001).
Walsh, C. T. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 303, 1805–1810 (2004).
Fischbach, M. A. & Walsh, C. T. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 106, 3468–3496 (2006).
Sieber, S. A. & Marahiel, M. A. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem. Rev. 105, 715–738 (2005).
Du, L., Sánchez, C. & Shen, B. Hybrid peptide-polyketide natural products: biosynthesis and prospects toward engineering novel molecules. Metab. Eng. 3, 78–95 (2001).
Walsh, C. T. et al. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr. Opin. Chem. Biol 5, 525–534 (2001).
Rix, U., Fischer, C., Remsing, L. L. & Rohr, J. Modification of post-PKS tailoring steps through combinatorial biosynthesis. Nat. Prod. Rep. 19, 542–580 (2002).
Yin, X., O'Hare, T., Gould, S. J. & Zabriskie, T. M. Identification and cloning of genes encoding viomycin biosynthesis from Streptomyces vinaceus and evidence for involvement of a rare oxygenase. Gene 312, 215–224 (2003).
Walsh, C. T. The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 41, 4–10 (2008).
Olano, C., Méndez, C. & Salas, J. A. Post-PKS tailoring steps in natural product-producing actinomycetes from the perspective of combinatorial biosynthesis. Nat. Prod. Rep. 27, 571–616 (2010).
Schobert, R. & Schlenk, A. Tetramic and tetronic acids: an update on new derivatives and biological aspects. Bioorg. Med. Chem. 16, 4203–4221 (2008).
Sánchez-Hidalgo, M., Núñez, L. E., Méndez, C. & Salas, J. A. Involvement of the beta subunit of RNA polymerase in resistance to streptolydigin and streptovaricin in the producer organisms Streptomyces lydicus and Streptomyces spectabilis. Antimicrob. Agents Chemother 54, 1684–1692 (2010).
Olano, C. et al. Deciphering biosynthesis of the RNA polymerase inhibitor streptolydigin and generation of glycosylated derivatives. Chem. Biol 16, 1031–1044 (2009).
Horna, D. H. et al. Biosynthesis of the RNA polymerase inhibitor streptolydigin in Streptomyces lydicus: tailoring modification of 3-methyl-aspartate. J. Bacteriol 193, 2647–2651 (2011).
Gómez, C. et al. Amino acid precursor supply in the biosynthesis of the RNA polymerase inhibitor streptolydigin by Streptomyces lydicus. J. Bacteriol 193, 4214–4223 (2011).
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. Practical Streptomyces Genetics, The John Innes Foundation: Norwich, (2000).
Fernández, E. et al. Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol 180, 4929–4937 (1998).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning: a laboratory manual, Cold Spring Harbour Laboratory press New York, Cold Spring Harbour, (1989).
Bierman, M. et al. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49 (1992).
Olano, C. et al. Biosynthesis of the angiogenesis inhibitor borrelidin by Streptomyces parvulus Tü4055: cluster analysis and assignment of functions. Chem. Biol 11, 87–97 (2004).
Menéndez, N. et al. Deoxysugar transfer during chromomycin A3 biosynthesis in Streptomyces griseus subsp. griseus: new derivatives with antitumor activity. Appl. Environ. Microbiol 72, 167–177 (2006).
Tang, G. L., Cheng, Y. Q. & Shen, B. Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem. Biol. 11, 33–45 (2004).
Garneau, S., Dorrestein, P. C., Kelleher, N. L. & Walsh, C. T. Characterization of the formation of the pyrrole moiety during clorobiocin and coumermycin A1 biosynthesis. Biochemistry 44, 2770–2780 (2005).
Bihlmaier, C. et al. Biosynthetic gene cluster for the polyenoyltetramic acid α-lipomycin. Antimicrob. Agents Chemother 50, 2113–2121 (2006).
Carlson, J. C. et al. Identification of the tirandamycin biosynthetic gene cluster from Streptomyces sp. 307-9. ChemBioChem 11, 564–572 (2010).
Mo, X. et al. Cloning and characterization of the biosynthetic gene cluster of the bacterial RNA polymerase inhibitor tirandamycin from marine-derived Streptomyces sp. SCSIO1666. Biochem. Biophys. Res. Commun 406, 341–347 (2011).
Du, L., Sánchez, C., Chen, M., Edwards, D. J. & Shen, B. The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem. Biol 7, 623–642 (2000).
Weber, T. et al. Molecular analysis of the kirromycin biosynthetic gene cluster revealed β-alanine as precursor of the pyridone moiety. Chem. Biol 15, 175–188 (2008).
Mendes, M. V., Antón, N., Martín, J. F. & Aparicio, J. F. Characterization of the polyene macrolide P450 epoxidase from Streptomyces natalensis that converts de-epoxypimaricin into pimaricin. Biochem. J. 386, 57–62 (2005).
Shah, S. et al. Cloning, characterization and heterologous expression of a polyketide synthase and P-450 oxidase involved in the biosynthesis of the antibiotic oleandomycin. J. Antibiot 53, 502–508 (2000).
Schultz, A. W. et al. Biosynthesis and structures of cyclomarins and cyclomarazines, prenylated cyclic peptides of marine actinobacterial origin. J. Am. Chem. Soc 130, 4507–4516 (2008).
Yu, T. W. et al. Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc. Natl Acad. Sci. USA 96, 9051–9056 (1999).
Doi-Katayama, Y. et al. Thioesterases and the premature termination of polyketide chain elongation in rifamycin B biosynthesis by Amycolatopsis mediterranei S699. J. Antibiot 53, 484–495 (2000).
Xu, J., Wan, E., Kim, C. J., Floss, H. G. & Mahmud, T. Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei S699. Microbiology 151, 2515–2528 (2005).
Sun, Y. et al. ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 148, 361–371 (2002).
Ou, X., Zhang, B., Zhang, L., Zhao, G. & Ding, X. Characterization of rrdA, a TetR family protein gene involved in the regulation of secondary metabolism in Streptomyces coelicolor. Appl. Environ. Microbiol. 75, 2158–2165 (2009).
Blankenfeldt, W. et al. Structure and function of the phenazine biosynthetic protein PhzF from Pseudomonas fluorescens. Proc. Natl Acad. Sci. USA 101, 16431–16436 (2004).
Van Lanen, S. G., Lin, S. & Shen, B. Biosynthesis of the enediyne antitumor antibiotic C-1027 involves a new branching point in chorismate metabolism. Proc. Natl Acad. Sci. USA 105, 494–499 (2008).
Barker, J. L. & Frost, J. W. Microbial synthesis of p-hydroxybenzoic acid from glucose. Biotechnol. Bioeng. 76, 376–390 (2001).
Stadthagen, G. et al. p-Hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J. Biol. Chem. 280, 40699–40706 (2005).
He, J. & Hertweck, C. Biosynthetic origin of the rare nitroaryl moiety of the polyketide antibiotic aureothin: involvement of an unprecedented N-oxygenase. J. Am. Chem. Soc 126, 3694–3695 (2004).
Van Kuilenburg, A. B., Stroomer, A. E., Van Lenthe, H., Abeling, N. G. & Van Gennip, A. H. New insights in dihydropyrimidine dehydrogenase deficiency: a pivotal role for beta-aminoisobutyric acid? Biochem. J. 379, 119–124 (2004).
Beck, Z. Q., Burr, D.A. & Sherman, D. H. Characterization of the β-methylaspartate-α-decarboxylase (CrpG) from the cryptophycin biosynthetic pathway. ChemBioChem 8, 1373–1375 (2007).
Acknowledgements
We thank Luz Valero Rustarazo from the Laboratory of Proteomics CIPF (network ProteoRed) for the MS analysis. This research was supported by a grant of the Spanish Ministry of Science and Innovation (BFU2006–00404) and Red Temática de Investigación Cooperativa de Centros de Cáncer (Ministry of Health, ISCIII-RETIC RD06/0020/0026) to JAS. We thank Obra Social Cajastur for financial support to Carlos Olano and the Spanish Ministry of Science and Innovation for PhD student fellowship (FPI) to Cristina Gómez. We also thank the Spanish Ministry of Science and Innovation (SAF2008–01845) and the Centro de Investigación Príncipe Felipe for their economical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Gómez, C., Olano, C., Palomino-Schätzlein, M. et al. Novel compounds produced by Streptomyces lydicus NRRL 2433 engineered mutants altered in the biosynthesis of streptolydigin. J Antibiot 65, 341–348 (2012). https://doi.org/10.1038/ja.2012.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2012.37
Keywords
This article is cited by
-
The Combinatorial Biosynthesis of “Unnatural” Products with Polyketides
Transactions of Tianjin University (2018)
-
Roles of type II thioesterases and their application for secondary metabolite yield improvement
Applied Microbiology and Biotechnology (2014)
