
Abstract
Microbes have made a phenomenal contribution to the health and well-being of people throughout the world. In addition to producing many primary metabolites, such as amino acids, vitamins and nucleotides, they are capable of making secondary metabolites, which constitute half of the pharmaceuticals on the market today and provide agriculture with many essential products. This review centers on these beneficial secondary metabolites, the discovery of which goes back 80 years to the time when penicillin was discovered by Alexander Fleming.
Similar content being viewed by others

Introduction
Back in 1928, Alexander Fleming1 began the microbial drug era when he discovered in a Petri dish seeded with Staphylococcus aureus that a compound produced by a mold killed the bacteria. The mold, identified as Penicillium notatum, produced an active agent that was named penicillin. Later, penicillin was isolated as a yellow powder and used as a potent antibacterial compound during World War II. By using Fleming's method, other naturally occurring substances, such as chloramphenicol and streptomycin, were isolated. Naturally occurring antibiotics are produced by fermentation, an old technique that can be traced back almost 8000 years, initially for beverages and food production. Beer is one of the world's oldest beverages, produced from barley by fermentation, possibly dating back to the sixth millennium BC and recorded in the written history of ancient Egypt and Mesopotamia. Another old fermentation, used to initiate the koji process, was that of rice by Aspergillus oryzae. During the past 4000 years, Penicillium roqueforti has been utilized for cheese production, and for the past 3000 years soy sauce in Asia and bread in Egypt has represented examples of traditional fermentations.2
Natural products with industrial applications can be produced from primary or secondary metabolism of living organisms (plants, animals or microorganisms). Owing to technical improvements in screening programs, and separation and isolation techniques, the number of natural compounds discovered exceeds 1 million.3 Among them, 50–60% are produced by plants (alkaloids, flavonoids, terpenoids, steroids, carbohydrates, etc.) and 5% have a microbial origin. Of all the reported natural products, approximately 20–25% show biological activity, and of these approximately 10% have been obtained from microbes. Furthermore, from the 22 500 biologically active compounds that have been obtained so far from microbes, 45% are produced by actinomycetes, 38% by fungi and 17% by unicellular bacteria.3 The increasing role of microorganisms in the production of antibiotics and other drugs for treatment of serious diseases has been dramatic. However, the development of resistance in microbes and tumor cells has become a major problem and requires much research effort to combat it.
Chemically synthesized drugs originating from natural products
Drugs of natural origin have been classified as (i) original natural products, (ii) products derived or chemically synthesized from natural products or (iii) synthetic products based on natural product structures. Evidence of the importance of natural products in the discovery of leads for the development of drugs for the treatment of human diseases is provided by the fact that close to half of the best selling pharmaceuticals in 1991 were either natural products or their derivatives.4 In this regard, of the 25 top-selling drugs reported in 1997, 42% were natural products or their derivatives and of these, 67% were antibiotics. Today, the structures of around 140 000 secondary metabolites have been elucidated.
It is important to understand that many chemically synthesized drugs owe their origin to natural sources. Applications of chemically synthesized natural metabolites include the use of a natural product derived from plant salicyclic acid derivatives present in white willow, wintergreen and meadowsweet to relieve pain and suffering. Concoctions of these plants were administered by Hippocrates back in the year 500 BC, and even earlier in Egypt and Babylonia, for fever, pain and childbirth. Synthetic salicylates were produced initially by Bayer in 1874, and later in 1897, Arthur Eichengrun at Bayer discovered that an acetyl derivative (aspirin), reduced acidity, bad taste and stomach irritation. These plant-based systems continue to play an essential role in health care, and it has been estimated by the World Health Organization (WHO) that approximately 80% of the world's inhabitants rely mainly on traditional medicines for their primary health care.5
Other synthesized compounds originating from natural products include a nonapeptide, designated teprotide, which was isolated from the venom of the Brazilian pit viper Bothrops jararaca.6 This led to the design and synthesis of angiotensin-converting enzyme (ACE) inhibitors such as captopril, which was the first marketed, orally active ACE inhibitor.7 Enalapril, another ACE inhibitor used in the treatment of cardiovascular disease, was approved for marketing by the Food and Drug Administration (FDA) in 1985.6
The alkaloid quinine, the active constituent of the ‘fever tree’ Cinchona succirubra, has been known for centuries by South American Indians to control malaria. During the twentieth century, massive programs to synthesize quinoline derivatives, based on the quinine prototype, were carried out. The first of the new quinolones to be used clinically as an antibacterial agent was nalidixic acid, which emerged as part of a large chemical synthesis program developed at the Sterling Winthrop Research Institute.8, 9 The program was begun when 7-chloro-1,4-dihydro-1-ethyl-4-oxoquinolone-3-carboxylic acid was obtained as a side product during purification of chloroquine and found to have antibacterial activity. The best compound found in the program was nalidixic acid, which had remarkable activity against Gram-negative bacteria and was shown to be an inhibitor of DNA gyrase. Its discovery led to a whole series of synthetic quinolone and fluoroquinolone antibiotics (pefloxacin, norfloxacin, ciprofloxacin, levofloxacin, ofloxacin, lomefloxacin, sparfloxacin, etc.), which have been very successful in medicine and have achieved major commercial success (Table 1). It is important to appreciate that all quinolones, though synthetic, are based on the structure of the natural plant product quinine.
Secondary metabolites have exerted a major impact on the control of infectious diseases and other medical conditions, and the development of pharmaceutical industry. Their use has contributed to an increase in the average life expectancy in the USA, which increased from 47 years in 1900 to 74 years (in men) and 80 years (in women) in 2000.11 Probably, the most important use of secondary metabolites has been as anti-infective drugs. In 2000, the market for such anti-infectives was US$55 billion (Table 1) and in 2007 it was US$66 billion.
Table 1 shows that, among the anti-infective drugs, antivirals represent more than 20% of the market. Two antivirals that are chemically synthesized today were originally isolated from marine organisms. They are acyclovir (active against the herpes virus by inhibition and inactivation of DNA polymerase) and cytarabine (active against non-Hodgkin's lymphoma). Both compounds are nucleoside analog drugs, originally isolated from sponges.12 Other antiviral applications of natural compounds are related to human immunodeficiency virus (HIV) treatment. In the pathogenesis of this disease, HIV-1, similar to other retroviruses, depends on its stable integration into the host genome to facilitate efficient replication of the viral RNA and maintenance of the infected state. Therefore, de novo viral DNA synthesized during reverse transcription is immediately integrated into the host cell DNA (through the integration step), allowing for further transcription of viral RNA. In the late phase of HIV viral replication, the large precursor polyprotein (gag-pol precursor, Pr 160) must be appropriately cleaved by a viral protease. The cleavage of the gag precursor protein of HIV is critical for the maturation and infectivity of the viral particle. Without the appropriate cleavage of the precursor polyproteins, non-infectious viral particles are generally produced. To confront this problem, a tremendous effort has been made at the US National Cancer Institute (NCI), in search of natural metabolites capable of inhibiting HIV reverse transcriptase and HIV protease. Chemically synthesized derivatives of these compounds are the main agents now used against HIV. Furthermore, reports have been published on natural product inhibitors of HIV integrase obtained from among the marine ascidian alkaloids; that is, the lamellarins (produced by the mollusk Lamellaria sp.), and from terrestrial plants (Baccharis genistelloides and Achyrocline satureioides). The most consistent anti-HIV activity was observed with extracts prepared from several Baccharis species.13 In addition, NCI has been evaluating the HIV-1 inhibitory activity of pepstatin A, a small pentapeptide produced by several Streptomyces species. It contains a unique hydroxyamino acid, statine, that sterically blocks the active site of HIV-1 protease.14, 15
Reasons for developing new antibiotics
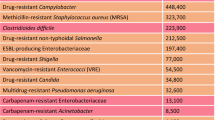
New antibiotics that are active against resistant bacteria are required. Bacteria have lived on the Earth for several billion years. During this time, they encountered in nature a wide range of naturally occurring antibiotics. To survive, bacteria developed antibiotic resistance mechanisms. Therefore, it is not surprising that they have become resistant to most of the natural antimicrobial agents that have been developed over the past 50 years.16 This resistance increasingly limits the effectiveness of current antimicrobial drugs. The problem is not just antibiotic resistance but also multidrug resistance. In 2004, more than 70% of pathogenic bacteria were estimated to be resistant to at least one of the currently available antibiotics.17 The so-called ‘superbugs’ (organisms that are resistant to most of the clinically used antibiotics) are emerging at a rapid rate. S. aureus, which is resistant to methicillin, is responsible for many cases of infections each year. The incidence of multidrug-resistant pathogenic bacteria is increasing. The Infectious Disease Society of America (IDSA) reported in 2004 that in US hospitals alone, around 2 million people acquire bacterial infections each year (http://www.idsociety.org/Content.aspx?id=4682). S. aureus is responsible for half of the hospital-associated infections and takes the lives of approximately 100 000 patients each year in the USA alone.18 The bacteria produce a biofilm in which they are encased and protected from the environment. Biofilms can grow on wounds, scar tissues and medical implants or devices, such as joint prostheses, spinal instrumentations, catheters, vascular prosthetic grafts and heart valves. More than 70% of the bacterial species producing such biofilms are likely to be resistant to at least one of the drugs commonly used in anti-infectious therapy.14 In hospitals, there are also other examples of Gram-positive (Enterococcus and Streptococcus) and Gram-negative pathogens (Klebsiella, Escherichia, Enterobacter, Serratia, Citrobacter, Salmonella and Pseudomonas); these hospital-inhabiting microbes are called ‘nosocomial bacteria.’ More than 60% of sepsis cases in hospitals are caused by Gram-negative bacteria.14 Among them, Pseudomonas aeruginosa accounts for almost 80% of these opportunistic infections. They represent a serious problem in patients hospitalized with cancer, cystic fibrosis and burns, causing death in 50% of cases. Other infections caused by Pseudomonas species include endocarditis, pneumonia and infections of the urinary tract, central nervous system, wounds, eyes, ears, skin and musculoskeletal system. This bacterium is another example of a natural multidrug-resistant microorganism. Although many strains are susceptible to gentamicin, tobramycin and amikacin, resistant forms have also developed. These multidrug-resistant bacteria make hospitals “dangerous places to be, especially if you are sick, but even if not.”19
Although we are seeing a steady increase in resistance in almost every pathogen to most of the current antibiotics over time, not all the antibacterial agents show the same rate of resistance development. For example, antimicrobials such as rifampicin, which targets single enzymes, are most susceptible to the development of resistance, whereas agents that inactivate several targets irreversibly generate resistance more slowly.
In addition to the antibiotic-resistance problem, new families of anti-infective compounds are needed to enter the marketplace at regular intervals to tackle the new diseases caused by evolving pathogens. At least 30 new diseases emerged in the 1980s and 1990s and they are growing in incidence. Emerging infectious organisms often encounter hosts with no prior exposure to them and thus represent a novel challenge to the host's immune system. Several viruses responsible for human epidemics have made a transition from animal host to humans and are now transmitted from human to human. HIV, responsible for the acquired immunodeficiency syndrome (AIDS) epidemic, is one example. Although it has not been proven, it is suspected that severe acute respiratory syndrome (SARS), caused by the SARS coronavirus, also evolved from a different species.20
In the early 1990s, after decades of decline, the incidence of tuberculosis began to increase. The epidemic took place owing to inadequate treatment regimens, a diminished public health system and the onset of the HIV/AIDS epidemic. The WHO has predicted that between 2000 and 2020, nearly 1 billion people will become infected with Mycobacterium tuberculosis and that this disease will cost the lives of 35 million people.
Sexually transmitted diseases have also increased during these decades, especially in young people (aged 15–24 years). The human papillomavirus, chlamydia, genital herpes, gonorrhea and HIV/AIDS are examples. HIV/AIDS has infected more than 40 million people in the world. Together with other diseases such as tuberculosis and malaria, HIV/AIDS accounts for over 300 million illnesses and more than 5 million deaths each year.
Additional evolving pathogens include the (i) Ebola virus, which causes the viral hemorrhagic fever syndrome with a resultant mortality rate of 88%; (ii) the bacterium Legionella pneumophila, a ubiquitous aquatic organism that lives in warm environments, which causes Legionnaire's disease, a pulmonary infection; (iii) the Hantavirus, which can infect humans with two serious illnesses: hemorrhagic fever with renal syndrome and Hantavirus pulmonary syndrome; (iv) at least three species of bacteria from the genus Borrelia, which cause Lyme disease, an emerging infection. In this case, the infection is acquired from the bite of ticks belonging to several species of the genus Ixodes. Borrelia burgdorferi is the predominant cause of Lyme disease in the US, whereas Borrelia afzelii and Borrelia garinii are implicated in most European cases. The disease presentation varies widely, and may include a rash and flu-like symptoms in its initial stage, followed by musculoskeletal, arthritic, neurologic, psychiatric and cardiac manifestations. In the majority of cases, symptoms can be eliminated with antibiotics, especially if the treatment begins early in the course of illness. However, late or inadequate treatment can lead to ‘late-stage’ Lyme disease that can be disabling and difficult to treat.21 (v) Other evolving pathogens include the Escherichia coli 0157:H7 (enterohemorrhagic E. coli), a strain that causes colitis and bloody diarrhea by producing a toxin called Shiga toxin, which damages the intestines. It is estimated that this bacterium causes infection in more than 70 000 patients a year in the USA. Another example is (vi) Cryptosporidium, an obligate intracellular parasite commonly found in lakes and rivers. Cryptosporidium parvum is one of the common species affecting the digestive and respiratory organs. Intestinal cryptosporidiosis is characterized by severe watery diarrhea. Pulmonary and tracheal cryptosporidiosis in humans is associated with coughing and is frequently a low-grade fever. People with severely weakened immune systems are likely to have more severe and more persistent symptoms than healthy individuals.
In the developing world, nearly 90% of the infectious disease deaths are caused by six diseases or disease processes: acute respiratory infections, diarrhea, tuberculosis, HIV, measles and malaria. In both the developing and developed nations, the leading cause of death by a wide margin is acute respiratory disease. In the developing world, acute respiratory infections are attributed primarily to seven bacteria: Bordetella pertussis, Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Mycoplasma pneumoniae, Chlamydophila pneumoniae and Chlamydia trachomatis. In addition, the major viral causes of respiratory infections include respiratory syncytial virus, human parainfluenza viruses 1 and 3, influenza viruses A and B, as well as some adenoviruses. These diseases are highly destructive in economic and social as well as in human terms and cause approximately 17 million deaths per year, and innumerable serious illnesses besides affecting the economic growth, development and prosperity of human societies.22 Morse23 identified six general factors in the emergence of infectious diseases: ecological changes, human demographics and behavior, international travel, technology and industry, microbial adaptation and change, and breakdown in public health measures.24
One additional reason for developing new antibiotics is related to their own toxicity. As with other therapeutic agents, the use of antibiotics may also cause side effects in patients. These include mild reactions such as upset stomach, vomiting and diarrhea (cephalosporins, macrolides, penicillins and tetracyclines), rash and other mild and severe allergic reactions (cephalosporins and penicillins), sensitivity to sunlight (tetracyclines), nervousness, tremors and seizures (quinolones). Some side effects are more severe and, depending on the antibiotic, may disrupt the hearing function (aminoglycosides), kidneys (aminoglycosides and polypeptides) or liver (rifampin).
Moves against antibiotic resistance development in bacteria
During recent decades, we have seen an increasing number of reports on the progressive development of bacterial resistance to almost all available antimicrobial agents. In the 1970s, the major problem was the multidrug resistance of Gram-negative bacteria, but later in the 1980s the Gram-positive bacteria became important, including methicillin-resistant staphylococci, penicillin-resistant pneumococci and vancomycin-resistant enterococci.25 In the past, the solution to the problem has depended primarily on the development of novel antimicrobial agents. However, the number of new classes of antimicrobial agents being developed has decreased dramatically in recent years.
The advent of resistant Gram-positive bacteria has been noticed by the pharmaceutical, biotechnology and academic communities. Some of these groups are making concerted efforts to find novel antimicrobial agents to meet this need. A new glycopeptide antibiotic, teicoplanin, was developed against infections with resistant Gram-positive bacteria, especially bacteria resistant to the glycopeptide vancomycin. In another instance, the approach involved the redesign of a mixture of two compounds, called streptogramin, into a new mixture, called pristinamycin, to allow administration of the drug parenterally and in higher doses than the earlier oral preparation.26 The two components of streptogramin, quinupristin and dalfopristin, were chemically modified to allow intravenous administration. The new combination, pristinamycin, was approved by the FDA for use against infections caused by vancomycin-resistant Enterococcus faecium.
Additional moves against resistant microorganisms are the glycylcyclines developed to treat tetracycline-resistant bacteria. These modified tetracyclines show potent activity against a broad spectrum of Gram-positive and Gram-negative bacteria, including strains that carry the two major tetracycline-resistance determinants, involving efflux and ribosomal protection. Two of the glycylcyline derivatives, DMG-MINO and DMG-DMDOT, have been tested against a large number of clinical pathogens isolated from various sources. The spectrum of activity of these compounds includes organisms with resistance to antibiotics other than tetracyclines; for example, methicillin-resistant staphylococci, penicillin-resistant S. pneumoniae and vancomycin-resistant enterococci.27 Tigecycline was approved by the FDA in 2005 as an injectable antibiotic.28
Among the novel class of antimicrobial agents used in treating resistance to Gram-positive infections, we can also mention the cyclic lipopeptide antibiotic daptomycin produced by Streptomyces roseosporus. This compound was approved in 2003 by the FDA for skin infections resulting from complications following surgery, diabetic foot ulcers and burns. It represents the first new natural antibiotic approved in many years. Its mode of action is distinct from any other approved antibiotic: it rapidly kills Gram-positive bacteria by disrupting multiple aspects of bacterial membrane function (by binding irreversibly to the bacterial cell membrane, causing membrane depolarization, destroying the ion concentration gradient and provoking the efflux of K+). It acts against most clinically relevant Gram-positive bacteria (Staphylococcus aureus, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae subsp. equisimilis and Enterococcus faecalis), and retains in vitro potency against isolates resistant to methicillin, vancomycin and linezolid. Traditionally, these infections were treated with penicillin and cephalosporins, but resistance to these agents became widespread.29, 30, 31, 32 Daptomycin seems to have a favorable side effect profile, and it might be used to treat patients who cannot tolerate other antibiotics.
Telithromycin, a macrolide antibiotic, is the first orally active compound of a new family of antibacterials named the ketolides. It shows potent activity against pathogens implicated in community-acquired respiratory tract infections, irrespective of their β-lactam, macrolide or fluoroquinolone susceptibility. Some of the microorganisms susceptible to this antibiotic are pneumococci, H. influenzae and Moraxella catarrhalis, including β-lactamase-positive strains. In addition, telithromycin has a very low potential for selection of resistant isolates or induction of cross-resistance found with other macrolides.33
Clavulanic acid, first detected in Streptomyces clavuligerus, contains a bicyclic β-lactam ring fused to an oxazolidine ring with an oxygen in place of a sulfur, a β-hydroxyethylidene substituent at C-2 and no acylamino group at C-6. It was first described in 1976 and shown to be a potent inhibitor of the β-lactamases produced by staphylococci and plasmid-mediated β-lactamases of E. coli, Klebsiella, Proteus, Shigella, Pseudomonas and Haemophilus. Although it is a broad-spectrum antibiotic, clavulanic acid possesses only very low antibacterial activity. Therefore, the molecule has been combined, as a β-lactamase inhibitor, with a variety of broad-spectrum semisynthetic penicillins. For example, when administered with amoxicillin, it is used for the treatment of infections caused by β-lactamase-producing pathogenic bacteria.34 It has world sales of over US$1 billion, and in 1995 it was the second largest selling antibacterial drug. Clavulanic acid can also be combined with ticarcillin, which is a penicillin effective against organisms such as E. coli, Proteus, Salmonella, Haemophilus, Pseudomonas and S. aureus. It is normally used in hospitals for treating severe infections affecting blood or internal organs, bones and joints, upper or lower airways or skin and soft tissue. The combination extends ticarcillin antimicrobial activity by inhibiting the action of the β-lactamases produced by certain bacteria.
Moves against resistance to antifungal agents
Mycosis is a condition in which fungi pass the resistance barriers of the human or animal body and establish infections. These organisms are harmless most of the time, but sometimes they can cause fungal infections. In most cases, these infections are not life threatening. However, when they are deeply invasive and disseminated, they lead to more serious infections, particularly in critically ill patients, elderly people and those who have conditions that affect the immune system (by disease or through the use of immunosuppressive agents). In addition, the use of antineoplastic and broad-spectrum antibiotics, prosthetic devices and grafts, and more aggressive surgery has increased invasive fungal infections. Patients with burns, neutropenia, pancreatitis or after organ transplantation (40% of liver transplants, 15–35% of heart transplants and 5% of kidney transplants) are also predisposed to fungal infection.35 Approximately 40% of death from nosocomial infections are caused by fungi, and 80% of these are caused by Candida and Aspergillus, although Cryptococcus spp., Fusarium spp., Scedosporium spp., Penicillium spp. and zygomycetes are increasingly involved.36 Pulmonary aspergillosis is the main factor involved in the death of recipients of bone marrow transplants, and Pneumocystis carinii is the leading cause of death in AIDS patients from Europe and North America.37
The rising incidence of invasive fungal infections and the emergence of broader fungal resistance have led to the need for novel antifungal agents. Amphotericin B is the first-line therapy for systemic infection because of its broad spectrum and fungicidal activity. However, considerable side effects limit its clinical utility. Echinocandins are large lipopeptide molecules that inhibit the synthesis of 1,3-β-D-glucan, a key component of the fungal cell wall. Three echinocandins (caspofungin, micafungin and anidulafungin) have reached the market. Caspofungin is also known as pneumocandin or MK-0991. This compound was the first cell-wall-active antifungal approved as a new injectable antifungal; this was in 2000.38 It irreversibly inhibits 1,3-β-D-glucan synthase, preventing the formation of glucan polymers and disrupting the integrity of fungal cell walls.39 It is more active and less toxic than amphotericin B and shows a broad spectrum of activity against Candida (including fluconozole resistance), Aspergillus, Histoplasma and P. carinii, the major cause of HIV death. Micafungin is licensed for clinical use in Asian countries and in the US. This compound exhibits extremely potent antifungal activity against clinically important fungi, including Aspergillus and azole-resistant strains of Candida. In animal studies, micafungin is as efficacious as amphotericin B with respect to improvement of survival rate. It is characterized by a linear pharmacokinetic profile and substantially fewer toxic effects. Anidulafungin is currently licensed in the US.40
Although several new antifungal drugs have been developed in the past 6 years, some patients remain resistant to treatments. The main reasons for this include intrinsic or acquired antifungal resistance, organ dysfunction preventing the use of some agents and drug interactions. In addition, some drugs penetrate poorly into sanctuary sites, including the eye and urine, and others are associated with considerable adverse events. However, there has been some progress. Posaconazole is a new member of the triazole class of antifungals. It has shown clinical efficacy in the treatment of oropharyngeal candidiasis and has potential as a salvage therapy for invasive aspergillosis, zygomycosis, cryptococcal meningitis and a variety of other fungal infections. It is available as an oral suspension and has a favorable toxicity profile. The wide spectrum of posaconazole activity in in vitro studies, animal models and preliminary clinical studies suggests that it represents an important addition to the antifungal armamentarium.41
Antibiotics wear many hats
In addition to the screening programs for antibacterial activity, the pharmaceutical industry has extended these programs to other disease areas.42, 43 Microorganisms are a prolific source of structurally diverse bioactive metabolites and have yielded some of the most important products of the pharmaceutical industry. Microbial secondary metabolites are now being used for applications other than antibacterial, antifungal and antiviral infections. For example, immunosuppressants have revolutionized medicine by facilitating organ transplantation.44 Other applications include antitumor drugs, enzyme inhibitors, gastrointestinal motor stimulator agents, hypocholesterolemic drugs, ruminant growth stimulants, insecticides, herbicides, coccidiostats, antiparasitics vs coccidia, helminths and other pharmacological activities. Further applications are possible in various areas of pharmacology and agriculture, developments catalyzed by the use of simple enzyme assays for screening before testing in intact animals or in the field.
Antitumor drugs
In the year 2000, approximately 10 million new cases of cancer were diagnosed in the world, resulting in 6 million cancer-related deaths. The tumor types with the highest incidence were lung (12.3%), breast (10.4%) and colorectal (9.4%).45
Microbial metabolites are among the most important of the cancer chemotherapeutic agents. They started to appear around 1940 with the discovery of actinomycin and since then many compounds with anticancer properties have been isolated from natural sources. More than 60% of the current compounds with antineoplasic activity were originally isolated as natural products or are their derivatives. Among the approved products deserving special attention are actinomycin D, anthracyclines (daunorubicin, doxorubicin, epirubicin, pirirubicin and valrubicin), bleomycin, mitosanes (mitomycin C), anthracenones (mithramycin, streptozotocin and pentostatin), enediynes (calcheamycin), taxol and epothilones.
Actinomycin D is the oldest microbial metabolite used in cancer therapy. Its relative, actinomycin A, was the first antibiotic isolated from actinomycetes. The latter was obtained from Actinomyces antibioticus (now Streptomyces antibioticus) by Waksman and Woodruff.46 As it binds DNA at the transcription initiation complex, it prevents elongation by RNA polymerase. This property, however, confers some human toxicity and it has been used primarily as an investigative tool in the development of molecular biology. Despite the toxicity, however, it has served well against Wilms tumor in children.
The anthracyclines are some of the most effective antitumor compounds developed, and are effective against more types of cancer than any other class of chemotherapy agents.47 They are used to treat a wide range of cancers, including leukemias, lymphomas, and breast, uterine, ovarian and lung cancers. Anthracyclines act by intercalating DNA strands, which result in a complex formation that inhibits the synthesis of DNA and RNA. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. In their cytotoxic effects, the binding to cell membranes and plasma proteins plays an important role. Their main adverse effects are heart damage (cardiotoxicity), which considerably limits their usefulness, and vomiting. The first anthracycline discovered was daunorubicin (daunomycin) in 1966, which is produced naturally by Streptomyces peucetius. Doxorubicin (adriamycin) was developed in 1967. Another anthracycline is epirubicin. This compound, approved by the FDA in 1999, is favored over doxorubicin in some chemotherapy regimens as it appears to cause fewer side effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar, which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas. Valrubicin is a semisynthetic analog of doxorubicin approved as a chemotherapeutic drug in 1999 and used to treat bladder cancer.
Bleomycin is a non-ribosomal glycopeptide microbial metabolite produced as a family of structurally related compounds by the bacterium Streptomyces verticillus. First reported by Umezawa et al.48 in 1966, bleomycin obtained FDA approval in 1973. When used as an anticancer agent (inducing DNA strand breaks), the chemotherapeutic forms are primarily bleomycins A2 and B2.
Mitosanes are composed of several mitomycins that are formed during the cultivation of Streptomyces caespitosus. Although the mitosanes are excellent antitumor agents, they have limited utility owing to their toxicity. Mitomycin C was approved by the FDA in 1974, showing activity against several types of cancer (lung, breast, bladder, anal, colorectal, head and neck), including melanomas and gastric or pancreatic neoplasms.49 Recently, mitomycin dimers have been explored as potential alternatives for lowering toxicity and increasing efficiency.50
Mithramycin (plicamycin) is an antitumor aromatic polyketide produced by Streptomyces argillaceous that shows antibacterial and antitumor activity.51 It is one of the older chemotherapy drugs used in the treatment of testicular cancer, disseminated neoplasms and hypercalcemia. It binds to G-C-rich DNA sequences, inhibiting the binding of transcription factors such as Sp1, which is believed to affect neuronal survival/death pathways. It may also indirectly regulate gene transcription by altering histone methylation. With repeated use, organotoxicity (kidney, liver and hematopoietic system) can become a problem.
Streptozotocin is a microbial metabolite with antitumor properties, produced by Streptomyces achromogenes. Chemically, it is a glucosamine-nitroso-urea compound. As with other alkylating agents in the nitroso-urea class, it is toxic to cells by causing damage to DNA, although other mechanisms may also contribute. The compound is selectively toxic to the β-cells of the pancreatic islets. It is similar enough to glucose to be transported into the cell by the glucose transport protein of these cells, but it is not recognized by the other glucose transporters. As β-cells have relatively high levels of glucose permease, the relative streptozotocin toxicity for these β-cells can be explained.52 In 1982, FDA granted approval for streptozotocin as a treatment for pancreatic islet cell cancer.
Pentostatin (deoxycoformycin) is an anticancer chemotherapeutic drug produced by S. antibioticus. It is classified as a purine analog, which mimics the nucleoside adenosine and thus tightly binds and inhibits adenosine deaminase (Ki of 2.5 × 10−12 M). It interferes with the cell's ability to process DNA.53 Pentostatin is commonly used to treat hairy cell leukemia, acute lymphocytic leukemia, prolymphocytic leukemia (B- and T-cell origin), T-cell leukemia and lymphoma. However, it can cause kidney, liver, lung and neurological toxicity.54 The FDA granted approval for pentostatin in 1993.
Calicheamicins are highly potent antitumor microbial metabolites of the enediyne family produced by Micromonospora echinospora. Their antitumor activity is apparently due to the cleavage of double-stranded DNA.55 They are highly toxic, but it was possible to introduce one such compound into the clinic by attaching it to an antibody that delivered it to certain cancer types selectively. This ingenious idea of the Wyeth Laboratories avoided the side effects of calicheamicin. In this regard, gemtuzumab is effective against acute myelogenous leukemia (AML). Calicheamicin is bound to a monoclonal antibody against a transmembrane receptor (CD33) expressed on cells of monocytic/myeloid lineage. CD33 is expressed in most leukemic blast cells, but in normal hematopoietic cells the intensity diminishes with maturation. It was approved by the FDA for use in patients over the age of 60 years with relapsed AML who are not considered candidates for standard chemotherapy.56
A successful non-actinomycete molecule is taxol (paclitaxel), which was first isolated from the Pacific yew tree, Taxus brevifolia, but is also produced by the endophytic fungi Taxomyces andreanae and Nodulisporium sylviforme.57 This compound inhibits rapidly dividing mammalian cancer cells by promoting tubulin polymerization and interfering with normal microtubule breakdown during cell division. The drug also inhibits several fungi (Pythium, Phytophthora and Aphanomyces) by the same mechanism. In 1992, taxol was approved for refractory ovarian cancer, and today it is used against breast and advanced forms of Kaposi's sarcoma.58 A new formulation is available in which paclitaxel is bound to albumin. Taxol sales amounted to US$1.6 billion in 2006 for Bristol Myers-Squibb, representing 10% of the company's pharmaceutical sales and its third largest selling product. Currently, taxol production uses plant cell fermentation technology.
The epothilones (a name derived from its molecular features: epoxide, thiazole and ketone) are macrolides originally isolated from the broth of the soil myxobacterium Sorangium cellulosum as weak agents against rust fungi.59 They were identified as microtubule-stabilizing drugs, acting in a similar manner to taxol.60, 61 However, they are generally 5–25 times more potent than taxol in inhibiting cell growth in cultures. Five analogs are now undergoing investigation as candidate anticancer drugs, and their preclinical studies have indicated a broad spectrum of antitumor activity, including taxol-resistant tumor cells. With the best currently available therapies, the median survival time for patients with metastatic breast cancer is only 2–3 years, and many patients develop resistance to taxanes or other chemotherapy drugs. One epothilone, ixabepilone, was approved in October 2007 by the FDA for use in the treatment of aggressive metastatic or locally advanced breast cancer no longer responding to currently available chemotherapies.62 In tumor cells, p-glycoprotein reduces intracellular antitumor drug concentrations, thereby limiting access of chemotherapeutic substrates to the site of action. The epothilones are attractive because they are active against p-glycoprotein-producing tumors and have good solubility.62 Epothilone B is a 16-membered polyketide macrolactone with a methylthiazole group connected to the macrocycle by an olefinic bond.
Testicular cancer is the most common cancer diagnosis in men between the ages of 15 and 35 years, with approximately 8000 cases detected in the United States annually.63 The majority (95%) of testicular neoplasms are germ cell tumors, which are relatively uncommon carcinomas, accounting for only 1% of all male malignancies. Remarkable progress has been made in the medical treatment of advanced testicular cancer, with a substantial increase in cure rates from approximately 5% in the early 1970s to almost 90% today.64, 65 This cure rate is the highest of any solid tumor, and improved survival is primarily due to effective chemotherapy. A major advance in chemotherapy for testicular germ cell tumors was the introduction of cisplatin in the mid-1970s. Two chemotherapy regimens are effective for patients with a good testicular germ cell tumor prognosis: four cycles of etoposide and cisplatin or three cycles of bleomycin, etoposide and cisplatin.66 Of the latter three agents, bleomycin and etoposide are natural products.
Enzyme inhibitors
Enzyme inhibitors have received increasing attention as useful tools, not only for the study of enzyme structures and reaction mechanisms but also for potential utilization in medicine and agriculture. Several enzyme inhibitors with various industrial uses have been isolated from microbes.67 The most important are (1) clavulanic acid, the inhibitor of β-lactamases discussed above in the section ‘Moves against antibiotic resistance development in bacteria,’ and the statins, hypocholesterolemic drugs presented below in the section ‘Hypocholesterolemic drugs.’ Some of the common targets for other inhibitors are glucosidases, amylases, lipases, proteases and xanthine oxidase (XO).
Acarbose is a pseudotetrasaccharide made by Actinoplanes sp. SE50. It contains an aminocyclitol moiety, valienamine, which inhibits intestinal α-glucosidase and sucrase. This results in a decrease in starch breakdown in the intestine, which is useful in combating diabetes in humans.68
Amylase inhibitors are useful for the control of carbohydrate-dependent diseases, such as diabetes, obesity and hyperlipemia.69, 70 Amylase inhibitors are also known as starch blockers because they contain substances that prevent dietary starches from being absorbed by the body. The inhibitors may also be useful for weight loss, as some versions of amylase inhibitors do show potential for reducing carbohydrate absorption in humans.71, 72 The use of amylase inhibitors for the treatment of rumen acidosis has also been reported.73 Examples of microbial α-amylase inhibitors are paim, obtained from culture filtrates of Streptomyces corchorushii,74 and TAI-A, TAI-B, oligosaccharide compounds from Streptomyces calvus TM-521.75
Lipstatin is a pancreatic lipase inhibitor produced by Streptomyces toxytricini that is used to combat obesity and diabetes. It interferes with the gastrointestinal absorption of fat.76 The commercial product is tetrahydrolipstatin, which is also known as orlistat.
In the pathogenic processes of some diseases, such as emphysema, arthritis, pancreatitis, cancer and AIDS, protease inhibitors are potentially powerful tools for inactivating target proteases. Examples of microbial products include antipain, produced by Streptomyces yokosukaensis, leupeptin from Streptomyces roseochromogenes and chymostatin from Streptomyces hygroscopicus.70 Leupeptin is produced by more than 17 species of actinomycetes.67
XO catalyzes the oxidation of hypoxanthine to uric acid through xanthine. An excessive accumulation of uric acid in the blood, called hyperuricemia, causes gout.77 The inhibitors of XO decrease the uric acid levels, which result in an antihyperuricemic effect. A potent inhibitor of XO, hydroxyakalone, was purified from the fermentation broth of Agrobacterium aurantiacum sp. nov., a marine bacterial strain.78
Fungal products are also used as enzyme inhibitors against cancer, diabetes, poisonings, Alzheimer's disease, etc. The enzymes inhibited include acetylcholinesterase, protein kinase, tyrosine kinase, glycosidases and others.79
Immunosuppresants
Suppressor cells are critical in the regulation of the normal immune response. An individual's immune system is capable of distinguishing between native and foreign antigens and of mounting a response only against the latter. A major role has been established for suppressor T lymphocytes in this phenomenon. Suppressor cells also play a role in regulating the magnitude and duration of the specific antibody response to an antigenic challenge. Suppression of the immune response either by drugs or by radiation, to prevent the rejection of grafts or transplants or to control autoimmune diseases, is called immunosuppression.
A number of microbial compounds capable of suppressing the immune response have been discovered. Cyclosporin A was originally introduced as a narrow-spectrum antifungal peptide produced by the mold, Tolypocladium nivenum (originally classified as Trichoderma polysporum and later as Tolypocladium inflatum), by aerobic fermentation. Cyclosporins are a family of neutral, highly lipophilic, cyclic undecapeptides containing some unusual amino acids, synthesized by a non-ribosomal peptide synthetase, cyclosporin synthetase. Discovery of the immunosuppressive activity led to its use in heart, liver and kidney transplants and to the overwhelming success of the organ transplant field.80 Cyclosporin was approved for use in 1983. It is thought to bind to the cytosolic protein cyclophilin (immunophilin) of immunocompetent lymphocytes, especially T lymphocytes. This complex of cyclosporin and cyclophilin inhibits calcineurin, which under normal circumstances is responsible for activating the transcription of interleukin-2. It also inhibits lymphokine production and interleukin release and therefore leads to a reduced function of effector T cells. Sales of cyclosporin A have reached US$1.5 billion per year.
Other important transplant agents include sirolimus (rapamycin) and tacrolimus (FK506), which are produced by actinomycetes. Rapamycin is especially useful in kidney transplants as it lacks the nephrotoxicity seen with cyclosporin A and tacrolimus. It is a macrolide, first discovered in 1975 as a product of S. hygroscopicus, and was initially proposed as an antifungal agent. However, this was abandoned when it was discovered that it had potent immunosuppressive and antiproliferative properties. This compound binds to the immunophilin FK506-binding protein (FKBP12), and this binary complex interacts with the rapamycin-binding domain and inactivates a serine-threonine kinase termed the mammalian target of rapamycin. The latter is known to control proteins that regulate mRNA translation initiation and G1 progression.81 The antiproliferative effect of rapamycin has also been used in conjunction with coronary stents to prevent restenosis, which usually occurs after the treatment of coronary artery disease by balloon angioplasty. Rapamycin also shows promise in treating tuberous sclerosis complex (TSC), a congenital disorder that leaves sufferers prone to benign tumor growth in the brain, heart, kidneys, skin and other organs. In a study of rapamycin as a treatment for TSC, University of California, Los Angeles (UCLA) researchers observed a major improvement in mice regarding retardation related to autism.82
As rapamycin has poor aqueous solubility, some of its analogs, RAD001 (everolimus), CCI-799 (tensirolimus) and AP23573 (ARIAD), have been developed with improved pharmaceutical properties. Everolimus is currently used as an immunosuppressant to prevent the rejection of organ transplants. Although it does not have FDA approval in the USA, it is approved for use in Europe and Australia, and phase III trials are being conducted in the US. Everolimus may have a role in heart transplantation as it has been shown to reduce chronic allograft vasculopathy in such transplants.83 Everolimus is also used in drug-eluting coronary stents as an immunosuppressant to prevent rejection. CCI-779 is a rapamycin ester that can be converted to rapamycin in vivo. RAD001 is a rapamycin analog currently being investigated in phase II trials for recurrent endometrial cancer as a single agent, and in phase I/II trials for the treatment of glioblastoma in combination with the inhibitor of certain epidermal growth factor receptor and vascular endothelial growth factor receptor family members.84 AP23573 is a novel non-prodrug rapamycin analog with a nonlinear pharmacokinetic behavior that has demonstrated antiproliferative activity against several human tumor cell lines in vitro and against experimental tumors in vivo.85 This agent is currently under evaluation in phase I–II trials, including patients with different tumors. Two additional small-molecule rapamycin analogs, AP23841 and AP23675, are currently in preclinical development for the treatment of bone metastases and primary bone cancer.86
Tacrolimus (FK506) was discovered in 1987 in Japan.87 It is produced by Streptomyces tsukubaensis. However, its use was almost abandoned because of dose-associated toxicity. Dr Thomas Starzl (University of Pittsburgh) rescued it by using lower doses, realizing that it was approximately 100 times more active as an immunosuppressive than cyclosporin A.88 It was introduced in Japan in 1993, and in 1994 it was approved by the FDA for use as an immunosuppressant in liver transplantation. Furthermore, its use has been extended to include bone marrow, cornea, heart, intestines, kidney, lung, pancreas, trachea, small bowel, skin and limb transplants, and for the prevention of graft-vs-host disease. Topically, it is also used against atopic dermatitis, a widespread skin disease. In the laboratory, tacrolimus inhibits the mixed lymphocyte reaction, the formation of interleukin-2 by T lymphocytes, and the formation of other soluble mediators, including interleukin-3 and interferon γ. Recently, it has been reported that tacrolimus inhibits tumor growth factor-β-induced signaling and collagen synthesis in human lung fibroblastic cells. This factor plays a pivotal role in tissue fibrosis, including pulmonary fibrosis. Therefore, tacrolimus may be useful for the treatment of pulmonary fibrosis, although its use in the acute inflammatory phase may exacerbate lung injury.89
Hypocholesterolemic drugs
Atherosclerosis is generally viewed as a chronic, progressive disease characterized by the continuous accumulation of atheromatous plaque within the arterial wall. The past two decades have witnessed the introduction of a variety of anti-atherosclerotic therapies. The statins form a class of hypolipidemic drugs used to lower cholesterol by inhibiting the enzyme HMG-CoA reductase, the rate-limiting enzyme of the mevalonate pathway of cholesterol biosynthesis. Inhibition of this enzyme in the liver stimulates low-density lipoprotein (LDL) receptors, resulting in an increased clearance of LDL from the bloodstream and a decrease in blood cholesterol levels. Through their cholesterol-lowering effect, they reduce the risk of cardiovascular disease, prevent stroke and reduce the development of peripheral vascular disease.90 In addition, they are anti-thrombotic and anti-inflammatory.
Currently there are a number of statins in clinical use. The entire group of statins reached an annual market of nearly US$30 billion before it became a generic pharmaceutical. The first member of the group (compactin; mevastatin) was isolated as an antibiotic product of Penicillium brevicompactum and later from Penicillium citrinum. Although not of commercial importance, compactin's derivatives achieved overwhelming medical and commercial success. An ethylated form, known as lovastatin (monacolin K; mevinolin), was isolated in the 1970s in the broths of Monascus ruber and Aspergillus terreus.91 Lovastatin, the first commercially marketed statin, was approved by the FDA in 1987. A semisynthetic derivative of lovastatin is simvastatin, a major hypocholesterolemic drug, selling for US$7 billion per year before becoming generic. Another statin, pravastatin (US$3.6 billion per year), is made through different biotransformation processes from compactin by Streptomyces carbophilus92 and Actinomadura sp.93 Other genera involved in the production of statins are Doratomyces, Eupenicillium, Gymnoascus, Hypomyces, Paecilomyces, Phoma, Trichoderma and Pleurotus.94 A synthetic compound, modeled from the structure of the natural statins, is atorvastin, which has been the leading drug of the entire pharmaceutical industry in terms of market share (approximately US$14 billion per year) for many years.
Insecticides
An insecticide is a pesticide used against insects in all developmental forms. They include ovicides and larvicides used against the eggs and larvae of insects, respectively. Insecticides are used in agriculture, medicine, industry and households. The use of insecticides is believed to be one of the major factors behind the increase in agricultural productivity in the twentieth century.
Synthetic insecticides pose some hazards, whereas natural insecticides offer adequate levels of pest control and pose fewer hazards. Microbially produced insecticides are especially valuable because their toxicity to non-target animals and humans is extremely low. Compared with other commonly used insecticides, they are safe for both the pesticide users and consumers of treated crops. The action of microbial insecticides is often specific to a single group or species of insects, and this specificity means that most microbial insecticides do not naturally affect beneficial insects (including predators or parasites of pests) in treated areas.
The spinosyns (A83543 group) are a group of natural products produced by Saccharopolyspora spinosa that were discovered in 1989. The researchers isolated spinosyn A and D, as well as 21 minor analogs. They are active on a wide variety of insect pests, especially lepidopterans and dipterans, but do not have antibiotic activity.95 The compounds attack the nervous system of insects by targeting two key neurotransmitter receptors, with no cross-resistance to other known insecticides. The spinosyns are a family of macrolides with 21 carbon atoms, containing four connected rings of carbon atoms at their core to which two deoxysugars (forosamine and 2,3,4, tri-O-methylrhamnose, which are required for bioactivity) are attached. Novel spinosyns have been prepared by biotransformation, using a genetically engineered strain of Saccharopolyspora erythraea.96 A mixture of spinosyn A (85%) and D (15%) (spinosad) is being produced through fermentation and was introduced to the market in 1997 for the control of chewing insects on a variety of crops. Spinosyn formulations were recently approved for use on organic crops and for animal health applications.
Recently, a new naturally occurring series of insect-active compounds was discovered from a novel soil isolate, Saccharopolyspora pogona NRRL30141. 97 The culture produced a unique family of over 30 new spinosyns. They have a butenyl substitution at the 21 position on the spinosyn lactone and are named butenyl-spinosyns or pogonins.
Herbicides
Herbicides are chemicals marketed to inhibit or interrupt normal plant growth and development. They are widely used in agriculture, industry and urban areas for weed management. Approximately 30 000 kinds of weeds are widely distributed in the world; yield losses caused by 1800 kinds of weeds are approximately 9.7% of total crop production every year.98 Herbicides provide cost-effective weed control with a minimum of labor. Most are used on crops planted in large acreages, such as soy, cotton, corn and canola.99 There are numerous classes of herbicides with different modes of action, as well as different potentials for adverse effects on health and the environment. Over the past century, chemical herbicides, used to control various weeds, may have caused many serious side effects, such as injured crops, threat to the applicator and others exposed to the chemicals, herbicide-resistant weed populations, reduction of soil and water quality, herbicide residues and detrimental effects on non-target organisms.100 For example, alachlor and atrazine were reported to cause cancer in animal tests. With increasing global environmental consciousness, bioherbicides, which are highly effective for weed control and environmentally friendly as well, are very attractive both for research and for application. Microbial herbicides can be divided into microbial preparations (microorganisms that control weeds) and microbially derived herbicides.
The first microbial herbicide was independently discovered in Germany and Japan. In 1972, the Zähner group in Germany isolated phosphinothricin tripeptide, a peptide antibiotic consisting of two molecules of L-alanine and one molecule of the unusual amino acid L-phosphinothricin; that is, N(4[hydroxyl(methyl)phosphinoyl]homoalanyl)alanylalanine. They isolated it from Streptomyces viridochromogenes as a broad-spectrum antibacterial including activity against Botrytis cinerea.101 In Japan, it was discovered at the Meiji Seiki laboratories in 1973 from S. hygroscopicus and named bialaphos.102 The bioactive L-phosphinothricin is a structural analog of glutamic acid, acting as a competitive inhibitor of glutamine synthetase, and has bactericidal (Gram-positive and Gram-negative bacteria), fungicidal (B. cinerea) and herbicidal properties.103 Glufosinate (DL-phosphinothricin) (without Ala-Ala) was developed as a herbicide. Therefore, the agent acts as a herbicide with or without Ala-Ala. Bialaphos has no influence on microorganisms in the soil and is easily degraded in the environment, having a half-life of only 2 h. This low level of environmental impact is of great interest to environmentalists.
Antiparasitics and ruminant growth stimulants
In 2006, the global animal health market was valued at US$16 billion, of which 29% was derived from parasiticides. Parasites are organisms that inhabit the body and benefit from a prolonged, close association with the host. Antiparasitics are compounds that inhibit the growth or reproduction of a parasite; some antiparasitics directly kill parasites. In general, parasites are much smaller than their hosts, show a high degree of specialization for their mode of life and reproduce more quickly and in greater numbers than their hosts. Classic examples of parasitism include the interactions between vertebrate hosts and such diverse animals as tapeworms, flukes, Plasmodium species and fleas. Parasitic infections can cause potentially serious health problems and even kill the host. Parasites mainly enter the body through the mouth, usually through ingestion of tainted food or drink. This is a very common problem in tropical areas, but is not limited to those regions. There are 3200 varieties of parasites in four major categories: Protozoa, Trematoda, Cestoda and Nematoda. The major groups include protozoans (organisms having only one cell) and parasitic worms (helminths). Each of these can infect the digestive tract, and sometimes two or more can cause infection at the same time. The WHO reported that approximately 25% of the world's population is infected with roundworms. In addition, a major agricultural problem has been the infection of farm animals by worms.
The predominant type of antiparasitic screening effort over the years was the testing of synthetic compounds against nematodes, and some commercial products did result. Certain antibiotics were also shown to possess antihelmintic activity against nematodes or cestodes, but these failed to compete with the synthetic compounds. Although Merck had earlier developed a commercially useful synthetic product, thiabendazole, they had enough foresight to examine microbial broths for antihelmintic activity, and found a non-toxic fermentation broth that killed the intestinal nematode Nematosporoides dubius in mice. The Streptomyces avermitilis culture, isolated by Ōmura and coworkers at the Kitasato Institute in Japan,104 produced a family of secondary metabolites (eight compounds) with both antihelmintic and insecticidal activities. These compounds, named ‘avermectins,’ are pentacyclic, 16-membered macrocyclic lactones, that harbor a disaccharide of the methylated sugar, oleandrose, with exceptional activity against parasites, especially Nemathelminthes (nematodes) and arthropod parasites (10 times higher than any known synthetic antihelmintic agent). Surprisingly, avermectins lack activity against bacteria and fungi, do not inhibit protein synthesis and are not ionophores. Instead, they interfere with neurotransmission in many invertebrates, causing paralysis and death by neuromuscular attacks.105
The annual market for avermectins surpasses US$1 billion. They are used against both nematode and arthropod parasites in sheep, cattle, dogs, horses and swine. A semisynthetic derivative, 22,23-dihydro-avermectin B1 (‘ivermectin’) is 1000 times more active than thiabendazole and is a commercial veterinary product. The efficacy of ivermectin has made it a promising candidate for the control of human onchocerciasis and human strongyloidiasis.106 Another avermectin, called doramectin (or cyclohexyl avermectin B1), produced by ‘mutational biosynthesis’ was commercialized for use by food animals.107 A semisynthetic monosaccharide derivative of doramectin called selamectin is the most recently commercialized avermectin, and is active against heartworms (Dirofilaria immitis) and fleas in companion animals. Although the macrocyclic backbone of each of these molecules (ivermectin, doramectin and selamectin) is identical, there are different substitutions at pharmacologically relevant sites such as C-5, C-13, C-22,23 and C-25.108 The avermectins are closely related to the milbemycins, a group of non-glycosidated macrolides produced by S. hygroscopicus subsp. Aureolacrimosus.109 These compounds possess activity against worms and insects.
Coccidiostats are used for the prevention of coccidiosis in both extensively and intensively reared poultry. Coccidiosis is the name given to a common intestinal disease caused by the invading protozoan parasites of the genus Eimeria that affects several different animal species (cattle, dogs, cats, poultry, etc.). The major damage is caused by the rapid multiplication of the parasite in the intestinal wall and the subsequent rupture of the cells of the intestinal lining, leading to high mortality and severe loss of productivity. Coccidia are obligate intracellular parasites that show host specificity; only cattle coccidia will cause disease in cattle; other species-specific coccidia will not.
For many years, synthetic compounds were used to combat coccidiosis in poultry; however, resistance developed rapidly. A solution came on the scene with the discovery of the narrow-spectrum polyether antibiotic monensin, which had extreme potency against the coccidian.110 Made by Streptomyces cinnamonensis, monensin led the way for additional microbial ionophoric antibiotics, such as lasalocid, narasin and salinomycin. All are produced by various Streptomyces species. They form complexes with the polar cations K+, Na+, Ca2+ and Mg2+, severely affecting the osmotic balance in the parasitic cells and thus causing their death.111 The widespread use of anticoccidials has revolutionized the poultry industry by reducing the mortality and production losses caused by coccidiosis. Of great interest was another extremely valuable application of monensin; that is, growth promotion in ruminants. Synthetic chemicals had been tested for years to inhibit wasteful methane production by cattle and sheep and increase fatty acid formation (especially propionate) to improve feed efficiency; however, they failed. The solution was monensin, which became a major success as a ruminant growth enhancer.110
For more than 40 years, certain antibiotics have been used in food-animal production to enhance feed utilization and weight gain.112 From a production standpoint, feed antibiotics have been consistently shown to improve animal weight gain and feed efficiency, especially in younger animals. These responses are probably derived from an inhibitory effect on the normal microbiota, which can lead to reduced intestinal inflammation and improved nutrient utilization.113 Pigs in the USA are exposed to a great variety of antibiotics. These include β-lactam antibiotics (including penicillins), lincosamides and macrolides (including erythromycin and tetracyclines). All these groups have members that are used to treat infections in humans. In addition, bacitracin, flavophospholipol, pleuromutilins, quinoxalines and virginiamycin are utilized as growth stimulants. Flavophospholipol and virginiamycin are also used as growth promoters in poultry.
As described above, cattle are also exposed to ionophores such as monensin to promote growth. The Animal Health Institute of America114 has estimated that without the use of growth-promoting antibiotics, the USA would require an additional 452 million chickens, 23 million more cattle and 12 million more pigs to reach the levels of production attained by the current practices.
Considering that animal health research and the development of new anti-infective product discovery have decreased, the discovery of new antibiotics has decreased over the past 15 years, with few new drug approvals.115 Therefore, it will be incumbent on veterinary practitioners to use the existing products in a responsible manner to ensure their longevity. It remains to be seen what effects the dearth of new antibiotics for veterinary medicine will have on the future practice of veterinary medicine, production agriculture, food safety and public health.116
Since the 1999 EU decision to prohibit antibiotic use for food-animal growth promotion, four antibiotic growth promoters have been banned, including the macrolide drugs tylosin and spiramycin.117 Although macrolides are no longer formally used as ‘growth promoters,’ their use under veterinary prescription has risen from 23 tons in 1998 to 55 tons in 2001, which suggests that more of them are being used now than before the prohibition.
Gastrointestinal motor stimulators
It is well known that the most effective route for feeding is via the gastrointestinal tract. Many critically ill patients who accept early feeding improve their health. In some post-operative patients, gastric stasis and excessive volumes in the stomach increase the risk of aspiration and subsequent pneumonia. On account of the importance of achieving early and adequate nutritional intake, it is common practice in many intensive care units to use drugs to improve gastrointestinal motility.
Erythromycin is a macrolide antibiotic with a broad spectrum of activity. It is well recognized that when prescribed, either intravenously or orally, it causes side effects, such as diarrhea, nausea and vomiting. These side effects are, in part, due to the action of erythromycin at motilin receptors in the gut. This makes this antibiotic very attractive to be used in ill patients with gastrointestinal motility problems. There have been some developments on erythromycin analogs that lack antibiotic action but retain action at motilin receptors. These have been named ‘motilides.’118, 119 Recently, an orally active erythromycin-derived motilin receptor agonist (mitemcinal) has been tested in patients with idiopathic and diabetic gastroparesis. In both cases, an improvement of gastroparetic symptoms was observed.120
Final comments
The 80-year contribution of microorganisms to medicine and agriculture has been overwhelming. However, antibiotic resistance in microbes has created a dangerous situation and the need for new antibiotics is clear. Unfortunately, most of the large pharmaceutical companies have abandoned the search for new antimicrobial compounds. Owing to the economics, they have concluded that drugs directed against chronic diseases offer a better revenue stream than do antimicrobial agents, as for the latter the length of treatment is short and government restriction is likely. Some small pharmaceutical and biotechnology companies are developing antibiotics, but most depend on venture capital rather than sales income, and with the present regulations, they face huge barriers to enter the market. These barriers were raised with the best intentions of ensuring public safety, but will have the opposite effect if they terminate antibiotic development while resistance continues to increase.121 However, there are some bright possibilities. One of the most promising is the utilization of uncultivated microorganisms. Considering that 99% of the bacteria and 95% of the fungi have not been cultivated in the laboratory, putting efforts into finding means to grow such microorganisms are proceeding and succeeding.122 Furthermore, researchers are now extracting bacterial DNA from soil and marine habitats, cloning large fragments into, for example, bacterial artificial chromosomes, expressing in a host bacterium and screening the library for new antibiotics. This metagenomic effort is allowing access to a vast untapped reservoir of genetic and metabolic diversity,123, 124 which could result in the discovery of new and useful natural products.125 In addition to these two relatively new techniques, the chemical and biological modification of old antibiotics could still supply new and powerful drugs. These comments also apply to non-antibiotics such as antitumor agents and other microbial products.
References
Fleming, A. On the antibacterial action of cultures of Penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 10, 226–236 (1929).
Hölker, U., Höfer, M. & Lenz, J. Biotechnological advantages of laboratory-scale solid-state fermentation with fungi. Appl. Microbiol. Biotechnol. 64, 175–186 (2004).
Berdy, J. Bioactive microbial metabolites. A personal view. J. Antibiot. 58, 1–26 (2005).
Cragg, G. M., Newman, D. J. & Snader, K. M. Natural products in drug discovery and development. J. Nat. Prod. 60, 52–60 (1997).
Farnsworth, N. R., Akerele, O., Bingel, A. S., Soejarto, D. D. & Guo, Z. Medicinal plants in therapy. Bull. WHO 63, 965–981 (1985).
Patchett, A. A. Natural products and design: interrelated approaches in drug discovery. J. Med. Chem. 45, 5609–5616 (2002).
Cushman, D. W. & Ondetti, M. A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 5, 1110–1112 (1999).
Overbye, K. M. & Barrett, J. F. Antibiotics: where did we go wrong? Drug Discov. Today 10, 45–52 (2005).
Topliss, J. G. et al. Natural and synthetic substances related to human health. Pure Appl. Chem. 74, 1957–1985 (2002).
Barber, M. S. The future of cephalosporins business. Chim. Oggi. 19, 9–13 (2001).
Lederberg, J. Infectious history. Science 288, 287–293 (2000).
Rayl, A. J. S. Oceans: medicine chests of the future? Scientist 13, 1–5 (1999).
Robinson, W. E., Reinecke, M. G., Abdel-Malek, S., Jia, O. & Chow, A. Inhibitors of HIV-1 replication that inhibit HIV integrase. Proc. Natl Acad. Sci. USA 93, 6326–6331 (1996).
Cragg, G. M. & Newman, D. J. Medicinals for the millennia. The historical record. Ann. NY Acad. Sci. 953a, 3–25 (2001).
Yang, S. S., Cragg, G. M., Newman, D. J. & Bader, J. P. Natural products based anti-HIV drug discovery and development facilitated by the NCI developmental therapeutics program. J. Nat. Prod. 64, 265–277 (2001).
Hancock, R. E. W. The end of an era? Nat. Rev. Drug Discov. 6, 26 (2007).
Katz, M. L., Mueller, L. V., Polyakov, M. & Weinstock, S. F. Where have all the antibiotic patents gone? Nat. Biotechnol. 24, 1529–1531 (2006).
Balaban, N. & Dell’Acqua, G. Barriers on the road to new antibiotics. Scientist 19, 42–43 (2005).
Levin, B. R. & Bonten, M. J. M. Cycling may be bad for your health. Proc. Natl Acad. Sci. USA 101, 13101–13102 (2004).
Ecker, D. J. et al. The Microbial Rosetta Stone Database: a compilation of global and emerging infectious microorganisms and bioterrorist threat agents. BMC Microbiol. 5, 19 (2005).
Ryan, K. J. & Ray, C. G. in Sherris Medical Microbiology 4th edn, 434–437 (McGraw Hill, New York, 2004).
OECD Forum. Health of Nations: Combating Infectious Diseases. Organisation for Economic Co-operation and Development. Centre de Conférences Internationales, 19 avenue Kléber 75116 Paris, France. 12–13 May 2004.
Morse, S. S. The public health threat of emerging viral disease. J. Nutr. 127, 951S–957S (1997).
Coates, A., Hu, Y., Bax, R. & Page, C. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov. 1, 895–910 (2002).
Moellering, R. C. Jr Problems with antimicrobial resistance in Gram-positive cocci. Clin. Infect Dis. 26, 1177–1178 (1998).
Bacque, E., Barriere, J. C. & Berthand, N. Recent progress in the field of antibacterial pristinamycins. Curr. Med. Chem. Anti-infect. Agents 4, 185–217 (2005).
Sum, P. E., Sum, F. W. & Projan, S. W. Recent developments in tetracycline antibiotics. Curr. Pharm. Des. 4, 119–132 (1998).
Sum, P. E. Case studies in current drug development: ‘glycylcyclines’. Curr. Opin. Chem. Biol. 10, 374–379 (2006).
Tally, F. P. & DeBruin, M. F. Development of daptomycin for Gram-positive infections. J. Antimicrob. Chemother. 46, 523–526 (2000).
Raja, A., LaBonte, J., Lebbos, J. & Kirkpatrick, P. Daptomycin. Nat. Rev. Drug Discov. 2, 943–944 (2003).
Eisenstein, B. I. Lipopeptides, focusing on daptomycin, for the treatment of Gram-positive infections. Expert Opin. Investig. Drugs 13, 1159–1169 (2004).
LaPlante, K. L. & Rybak, M. J. Daptomycin—a novel antibiotic against Gram-positive pathogens. Expert Opin. Pharmacother. 5, 2321–2331 (2004).
Leclercq, R. Overcoming antimicrobial resistance: profile of a new ketolide antibacterial, telithromycin. J. Antimicrob. Chemother. 48, 9–23 (2001).
Jensen, S. E. & Paradkar, A. S. Biosynthesis and molecular genetics of clavulanic acid. Antonie van Leeuwenhoek 75, 125–133 (1999).
Singh, N. Invasive aspergillosis in organ transplant recipients: new issues in epidemiologic characteristics, diagnosis, and management. Med. Mycol. 43 (suppl. 1), S267–S270 (2005).
Enoch, D. A., Ludlam, H. A. & Brown, N. M. Invasive fungal infections: a review of epidemiology and management options. J. Med. Microbiol. 55, 809–818 (2006).
Alexander, B. D. & Perfect, J. R. Antifungal resistance trends towards the year 2000. Implications for therapy and new approaches. Drugs 54, 657–678 (1997).
Hoang, A. Caspofungin acetate: an antifungal agent. Am. J. Health Syst. Pharm. 58, 1206–1217 (2001).
Georgopapadakou, N. H. Antifungals targeted to cell wall: focus on β-1,3-glucan synthase. Expert Opin. Invest. Drugs 10, 269–280 (2001).
Ikeda, F. et al. Role of micafungin in the antifungal armamentarium. Curr. Med. Chem: 14, 1263–1275 (2007).
Kwon, D. S. & Mylonakis, E. Posaconazole: a new broad-spectrum antifungal agent. Expert Opin. Pharmacother. 8, 1167–1178 (2007).
Cardenas, M. E., Sanfridson, A., Cutler, N. S. & Heitman, J. Signal-transduction cascades as targets for therapeutic intervention by natural products. Trends Biotechnol. 16, 427–433 (1998).
Kremer, L., Douglas, J. D., Baulard, A. R., Morehouse, C. & Guy, M. R. Thiolactomycin and related analogues as novel anti-mycobacterial agents targeting KasA and KasB condensing enzymes in Mycobacterium tuberculosis. J. Biol. Chem. 275, 16857–16864 (2000).
Verdine, G. L. The combinatorial chemistry of nature. Nature 384, 11–13 (1996).
Schwartsmann, G. et al. Anticancer drug discovery and development throughout the world. J. Clin. Oncol. 20 18S, 47s–59s (2002).
Waksman, S. A. & Woodruff, H. B. Actinomyces antibioticus, a new soil organism antagonistic to pathogenic and non-pathogenic bacteria. J. Bacteriol. 42, 231–249 (1941).
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G. & Gianni, L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229 (2004).
Umezawa, H., Maeda, K., Takeuchui, T. & Okami, Y. New antibiotics, bleomycin A and B. J. Antibiot. 19A, 200–209 (1966).
Schein, P. S., Macdonald, J. S., Hoth, D. F. & Wooley, P. V. The FAM (5-fluorouracil, adriamycin, mitomycin C) and SMF (streptozotocin, mitomycin C, 5-fluorouracil) chemotherapy regimens. in Mitomycin C: Current Status and New Developments (eds Carter, S.K. & Crooke, S.T.) 133–143 (Academic Press, New York, 1979).
Paz, M. M., Kumar, G. S., Glover, M., Waring, M. J. & Tomasz, M. Mitomycin dimers: polyfunctional cross-linkers of DNA. J. Med. Chem. 47, 3308–3319 (2004).
Fernández, E. et al. Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol. 180, 4929–4937 (1998).
Wang, Z. & Gleichmann, H. GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 47, 50–56 (1998).
Showalter, D. H. et al. Improved production of pentostatin and identification of fermentation cometabolites. J. Antibiot. 45, 1914–1918 (1992).
Dang, N. H. et al. Pentostatin in T-non-Hodgkin's lymphomas: efficacy and effect on CD26+ T lymphocytes. Oncol. Rep. 10, 1513–1518 (2003).
Walker, S., Landovitz, R., Ding, W. D., Ellestad, G. A. & Kahne, D. Cleavage behavior of calicheamicin gamma 1 and calicheamicin T. Proc. Natl Acad. Sci. USA 89, 4608–4612 (1992).
Bross, P. F. et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 7, 1490–1496 (2001).
Zhao, K., Zhou, D., Ping, W. & Ge, J. Study on the preparation and regeneration of protoplast from taxol-producing fungus Nodulisporium sylviforme. Nat. Sci 2, 52–59 (2004).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477 (2007).
Gerth, K., Bedorf, N. & Hofle, G. Epothilons A and B: antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria): production, physico-chemical and biological properties. J. Antibiot. 49, 560–563 (1996).
Bollag, D. M., McQueney, P. A. & Zhu, J. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 55, 2325–2333 (1995).
Kowalski, R. J., Giannakakou, P. & Hamel, E. Activities of the microtubule-stabilizing agents epothilones A and B with purified tubulin and in cells resistant to paclitaxel (Taxol). J. Biol. Chem. 272, 2534–2541 (1997).
Goodin, S., Kane, M. P. & Rubin, E. H. Epothilones: mechanism of action and biologic activity. J. Clin. Oncol. 22, 2015–2025 (2004).
Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J. & Thun, M. J. Cancer statistics, 2007. CA Cancer J. Clin. 57, 43–66 (2007).
Einhorn, L. H. Curing metastatic testicular cancer. Proc. Natl Acad. Sci. USA 99, 4592–4595 (2002).
Feldman, D. R., Bosl, G. J., Sheinfeld, J. & Motzer, R. J. Medical treatment of advanced testicular cancer. J. Am. Med. Assoc. 299, 672–684 (2008).
Culine, S. et al. Refining the optimal chemotherapy regimen for good-risk metastatic nonseminomatous germ-cell tumors: a randomized trial of the Genito-Urinary Group of the French Federation of Cancer Centers (GETUG T93BP). Ann. Oncol. 18, 917–924 (2007).
Umezawa, H. Enzyme Inhibitors of Microbial Origin (University of Tokyo Press, Tokyo, Japan, 1972, ).
Truscheit, E., Frommer, W., Junge, B., Müller, L., Schmidt, D. D. & Wingender, W. Chemistry and biochemistry of microbial α-glucosidase inhibitors. Angew. Chem. Intl Ed. Engl. 20, 744–761 (1981).
Boivin, M., Flourie, B., Rizza, R. A., Go, V. L. & DiMagno, E. P. Gastrointestinal and metabolic effects of amylase inhibition in diabetics. Gastroenterology 94, 387–394 (1988).
Imada, C. Enzyme inhibitors of marine microbial origin with pharmaceutical importance. Mar. Biotechnol. (NY) 6, 193–198 (2004).
Díaz, E., Aguirre, C. & Gotteland, M. Effect of an α- amylase inhibitor on body weight reduction in obese women. Rev. Chil. Nutr. 31, 306–317 (2004).
Boivin, M., Zinsmeister, A. R., Go, V. L. & DiMagno, E. P. Effect of a purified amylase inhibitor on carbohydrate metabolism after a mixed meal in healthy humans. Mayo Clin. Proc. 62, 249–255 (1987).
Banks, B. J., Haxell, M. A., Lunn, G., Pacey, M. S. & Roberts, L. R. Treatment of rumen acidosis with alpha-amylase inhibitors European Patent EP1157696 (2006).
Arai, M., Oouchi, N. & Murao, S. Inhibitory properties of an α-amylase inhibitor, paim, from Streptomyces corchorushii. Agric. Biol. Chem. 49, 987–991 (1985).
Kangouri, K., Namiki, S., Nagate, T., Sugita, K. & Ōmura, S. Novel amylase inhibitors United States Patent 4197292 (1980).
Weibel, E. K., Hadvary, P., Hochuli, E., Kupfer, E. & Lengsfeld, H. Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. J. Antibiot. 40, 1081–1085 (1987).
Borges, F., Fernandes, E. & Roleira, F. Progress towards the discovery of xanthine oxidase inhibitors. Curr. Med. Chem. 9, 195–217 (2002).
Izumida, H., Miki, W., Sano, H. & Endo, M. Agar plate method, a new assay for chitinase inhibitors using a chitin-degrading bacterium. J. Marine Biotechnol. 2, 163–166 (1995).
Paterson, R. R. M. Fungal enzyme inhibitors as pharmaceuticals, toxins and scourge of PCR. Curr. Enzyme Inhib. 4, 46–59 (2008).
Borel, J. F. History of the discovery of cyclosporin and of its early pharmacological development. Wien. Klin. Wochenschr. 114, 433–437 (2002).
Aggarwala, D., Fernandez, M. L. & Solimanb, G. A. Rapamycin, an mTOR inhibitor, disrupts triglyceride metabolism in guinea pigs. Metabolism 55, 794–802 (2006).
Ehninger, D. et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14, 843–848 (2008).
Eisen, H. J. et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N. Eng. J. Med. 349, 847–858 (2003).
Huang, S. & Houghton, P. J. Targeting mTOR signaling for cancer therapy. Curr. Opin. Pharmacol. 3, 371–377 (2003).
Dancey, J. E. Therapeutic targets: MTOR and related pathways. Cancer Biol. Ther. 5, 1065–1073 (2006).
Chen, Y.-J. Targeted mTOR in human gynecologic cancers. J. Cancer Mol. 3, 101–106 (2007).
Kino, T. et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. 1. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. 40, 1249–1255 (1987).
Jain, A. B. et al. Correlation of rejection episodes with FK 506 dosage, FK 506 level and steroid following primary orthotopic liver transplant. Transplant Proc. 23, 3023–3025 (1991).
Nagano, J. et al. Use of tacrolimus, a potent antifibrotic agent, in bleomycin-induced lung fibrosis. Eur. Respir. J. 27, 460–469 (2006).
Nicholls, S. J. et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. J. Am. Med. Assoc. 297, 499–508 (2007).
Alberts, A. W. et al. Mevinolin, a highly potent competitive inhibitor of hydroxylmethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl Acad. Sci. USA 77, 3957–3961 (1980).
Serizawa, N. & Matsuoka, T. A two-component-type cytochrome P-450 monooxygenase system in a prokaryote that catalyzes hydroxylation of ML-236B to pravastatin, a tissue-selective inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Biochim. Biophys. Acta. 1084, 35–40 (1991).
Peng, Y. & Demain, A. L. A new hydroxylase system in Actinomadura sp. cells converting compactin to pravastatin. J. Ind. Microbiol. Biotechnol. 20, 373–375 (1998).
Alarcon, J., Aguila, S., Arancibia-Avila, P., Fuentes, O., Zamorano-Ponce, E. & Hernandez, M. Production and purification of statins from Pleurotus ostreatus (Basidiomycetes) strains. Z. Naturforsch. 58, 62–64 (2003).
Kirst, H. A. et al. Evaluation and development of spinosyns to control ectoparasites on cattle and sheep. Curr. Top. Med. Chem. 2, 675–699 (2002).
Gaisser, S. et al. Engineered biosynthesis of novel spinosyns bearing altered deoxyhexose substituents. Chem. Commun. 21, 618–619 (2002).
Hahn, D. R., Gustafson, G., Waldron, C., Bullard, B., Jackson, J. D. & Mitchell, J. Butenyl-spinosyns, a natural example of genetic engineering of antibiotic biosynthetic genes. J. Ind. Microbiol. Biotechnol. 33, 94–104 (2006).
Li, Y., Sun, Z., Zhuang, X., Xu, L., Chen, S. & Li, M. Research progress on microbial herbicides. Crop Prot. 22, 247–252 (2003).
Randall, J. M. Weed control for the preservation of biological diversity. Weed Technol. 10, 370–383 (1996).
Kline, W. N. & Duquesnel, J. G. Control of problem vegetation: a key to ecosystem management. Down Earth 51, 20–28 (1996).
Bayer, E. et al. Metabolic products of microorganisms. 98. Phosphinothricin and phosphinothricyl-alanyl-analine. Helv. Chim. Acta. 55, 224–239 (1972).
Kondo, Y. et al. Studies on a new antibiotic, SF-1293. I. Isolation and physico-chemical and biological characterization of SF-1293 substances. Sci. Rep. Meiji Seika 13, 34–41 (1973).
Schwartz, D., Berger, S., Heinzelmann, E., Muschko, K., Welzel, K. & Wohlleben, W. Biosynthetic gene cluster of the herbicide phosphinothricin tripeptide from Streptomyces viridochromogenes Tu494. Appl. Environ. Microbiol. 70, 7093–7102 (2004).
Stapley, E. O. Avermectins, antiparasitic lactones produced by Streptomyces avermitilis isolated from a soil in Japan. in Trends in Antibiotic Research (eds Umezawa, H., Demain, A.L., Hata, R. & Hutchinson, C.R.) 154–170 (Japan Antibiotic Research Association, Tokyo, Japan, 1982).
Tooth, J. A., Davis, M. W. & Avery, L. A. avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. EMBO J. 16, 5867–5879 (1997).
Shikiya, K. et al. Efficacy of ivermectin against Strongyloides stercoralis in humans. Intern. Med. 31, 310–312 (1992).
Goudie, A. C. et al. Doramectin-A potent novel endectocide. Vet. Parasitol. 49, 5–15 (1993).
Michael, B., Meinke, P. T. & Shoop, W. L. Comparison of ivermectin, doramectin, selamectin, and eleven intermediates in a nematode larval development assay. J. Parasitol. 87, 692–696 (2002).
Mishima, H., Ide, J., Muramatsu, S. & Ono, M. Milbemycins, a new family of macrolide antibiotics. Structure determination of milbemycins D, E, F, G, H, J and K. J. Antibiot. 36, 980–990 (1983).
Westley, J. W. Polyether antibiotics: versatile carboxylic acid ionophores produced by Streptomyces. Adv. Appl. Microbiol. 22, 177–223 (1977).
Matabudul, D. K., Lumley, I. D. & Points, J. S. The determination of 5 anticoccidial drugs (nicarbazin, lasalocid, monensin, salinomycin and narasin) in animal livers and eggs by liquid chromatography linked with tandem mass spectrometry (LC-MS-MS). Analyst 127, 760–768 (2002).
Page, S. W. The Role of Enteric Antibiotics in Livestock Production (National Association for Crop Production and Animal Health, Avcare, Australia, 2003).
Gaskins, H. R., Collier, C. T. & Anderson, D. B. Antibiotics as growth promotants: mode of action. Anim. Biotechnol. 13, 29–42 (2002).
Animal Health Institute USA. Antibiotic resistance back in the news. AHI Q 19, 1–4 (1998).
Nelson, R. Antibiotic development pipeline runs dry. Lancet 362, 1726–1727 (2003).
Shryock, T. R. The future of anti-infective products in animal health. Nat. Rev. Microbiol. 2, 425–430 (2004).
Casewell, M., Friis, C., Marco, E., McMullin, P. & Phillips, I. The European ban on growth-promoting antibiotics and emerging consequences for human and animal health. J. Antimicrob. Chemother. 52, 159–161 (2003).
Funabashi, Y. et al. Bioactive metabolites of EM574 and EM523, erythromycin derivatives having strong gastrointestinal motor stimulating activity. J. Antibiot. 49, 794–801 (1996).
Bradley, C. Erythromycin as a gastrointestinal prokinetic agent. Intensive Crit. Care Nurs. 17, 117–119 (2001).
McCallum, R. W. & Cynshi, O. Clinical trial: effect of mitemcinal (a motilin agonist) on gastric emptying in patients with gastroparesis—a randomized, multicentre, placebo-controlled study. Aliment. Pharmacol. Ther. 26, 1121–1130 (2007).
Livermore, D. M. The need for new antibiotics. Clin. Microbiol. Infect 10, 1–9 (2004).
Kaeberlein, T., Lewis, K. & Epstein, S. S. Isolating ‘uncultivable’ microorganisms in pure culture in a simulated natural environment. Science 296, 1127–1129 (2002).
Colwell, R. R. Fulfilling the promise of biotechnology. Biotechnol. Adv. 20, 215–228 (2002).
Gaudilliere, B., Bernardelli, P. & Berna, P. in Annual Reports in Medicinal Chemistry Vol. 36 (ed. Doherty, A.M.) 293–318 (Academic Press, San Diego, USA, 2001).
Clardy, J., Fischbach, M. A. & Walsh, C. T. New antibiotics from bacterial natural products. Nat. Biotechnol. 24, 1541–1550 (2006).
Acknowledgements
We thank Beatriz Ruiz and Marco A Ortiz for their assistance during the development of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Demain, A., Sanchez, S. Microbial drug discovery: 80 years of progress. J Antibiot 62, 5–16 (2009). https://doi.org/10.1038/ja.2008.16
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2008.16
Keywords
This article is cited by
-
Plant Growth-Promoting and Arsenic Accumulation Reduction Effects of Two Endophytic Bacteria Isolated from Brassica napus
Journal of Plant Growth Regulation (2024)
-
Amamine, an isoquinoline alkaloid from the Kitasatospora sp. HGTA304
The Journal of Antibiotics (2023)
-
Rubiflavin G, photorubiflavin G, and photorubiflavin E: Novel pluramycin derivatives from Streptomyces sp. W2061 and their anticancer activity against breast cancer cells
The Journal of Antibiotics (2023)
-
Recent Progress in the Development of Droplet-based Microfluidic Technologies for Phenotypic Screening using Cell-cell Interactions
Biotechnology and Bioprocess Engineering (2023)
-
Rapid identification of pyoverdines of fluorescent Pseudomonas spp. by UHPLC-IM-MS
BioMetals (2023)
