
Abstract
Mealybugs (Insecta: Hemiptera: Pseudococcidae) maintain obligatory relationships with bacterial symbionts, which provide essential nutrients to their insect hosts. Most pseudococcinae mealybugs harbor a unique symbiosis setup with enlarged betaproteobacterial symbionts (‘Candidatus Tremblaya princeps’), which themselves contain gammaproteobacterial symbionts. Here we investigated the symbiosis of the manna mealybug, Trabutina mannipara, using a metagenomic approach. Phylogenetic analyses revealed that the intrabacterial symbiont of T. mannipara represents a novel lineage within the Gammaproteobacteria, for which we propose the tentative name ‘Candidatus Trabutinella endobia’. Combining our results with previous data available for the nested symbiosis of the citrus mealybug Planococcus citri, we show that synthesis of essential amino acids and vitamins and translation-related functions partition between the symbiotic partners in a highly similar manner in the two systems, despite the distinct evolutionary origin of the intrabacterial symbionts. Bacterial genes found in both mealybug genomes and complementing missing functions in both symbioses were likely integrated in ancestral mealybugs before T. mannipara and P. citri diversified. The high level of correspondence between the two mealybug systems and their highly intertwined metabolic pathways are unprecedented. Our work contributes to a better understanding of the only known intracellular symbiosis between two bacteria and suggests that the evolution of this unique symbiosis included the replacement of intrabacterial symbionts in ancestral mealybugs.
Similar content being viewed by others
Introduction
Mealybugs (Insecta: Hemiptera: Pseudococcidae) are phloem sap-feeding insects, which harbor a unique intrabacterial symbiotic system within their bacteriome, a large specialized organ in the abdomen. Bacteriocytes of most pseudococcinae mealybugs (Hardy et al., 2008) contain betaproteobacterial symbionts, which themselves host gammaproteobacterial symbionts within their cytoplasm (von Dohlen et al., 2001). Similar to other plant sap-feeding insects (Baumann, 2005; Moran et al., 2008), these bacterial symbionts provide essential amino acids (and vitamins) scarce in the phloem diet of the mealybugs (McCutcheon and von Dohlen, 2011). The monophyletic betaproteobacterial lineage, named Tremblaya, and the insect hosts show congruent phylogenies, indicating a single infection followed by long-term co-speciation in ancestral mealybugs (Fukatsu and Nikoh, 2000; Thao et al., 2002; Baumann and Baumann, 2005; Downie and Gullan, 2005; Gruwell et al., 2010). However, Tremblaya was likely infected by different enterobacterial precursors in different mealybug lineages, as suggested by phylogenetic analyses of the 16S and 23S ribosomal RNA genes of these secondary symbionts (Fukatsu and Nikoh, 2000; Thao et al., 2002). Thus the unique nested symbiosis of mealybugs, also referred to as ‘a bug in a bug in a bug’, likely emerged multiple times during evolution (Thao et al., 2002).
To date, genome sequences of the symbiont pair of the citrus mealybug Planococcus citri (‘Candidatus Tremblaya princeps’ and ‘Candidatus Moranella endobia’) (McCutcheon and von Dohlen, 2011; López-Madrigal et al., 2013) and the sole symbiont of the mealybug Phenacoccus avenae (‘Ca. T. phenacola’) that does not contain an intrabacterial symbiont have been determined (Husnik et al., 2013). These revealed an extreme reduction of Tremblaya genomes making them one of the smallest known (Moran and Bennett, 2014). More than one-fifth of the 139 kb chromosome of ‘Ca. T. phenacola’ in P. citri is devoted to essential amino-acid biosynthesis, which is the central role of the symbiosis. However, Tremblaya in P. citri has lost several genes even in functions key for the maintenance of the symbiosis (such as genes involved in translation and essential amino-acid synthesis), and its genome is still in the process of reduction. Its intrabacterial partner, Moranella is metabolically more versatile and can complement some of these missing functions, but operation of the symbiosis requires a substantial supply of metabolites and gene products from the mealybug host (McCutcheon and von Dohlen, 2011; Husnik et al., 2013; López-Madrigal et al., 2013).
Recent studies demonstrated a heterogeneous nature of contribution of the insect hosts in maintaining the symbiosis in aphids, psyllids and mealybugs, which is mediated by eukaryotic genes as well as genes of bacterial origin encoded in the insect genomes (Nikoh et al., 2010; Hansen and Moran, 2011; Husnik et al., 2013; Sloan et al., 2014). It was suggested that bacterial genes were acquired by P. citri through ancient horizontal gene transfer events from a surprisingly diverse set of putative facultative bacterial symbionts. Genes of bacterial origin encoded in the mealybug genome seem to be overexpressed in the bacteriome and can bypass key reactions, for instance, in essential amino-acid and vitamin synthesis (Husnik et al., 2013). Thus the P. citri symbiosis requires a complex interplay between the insect host and its two bacterial symbionts, which also involves gene products of horizontally acquired bacterial genes encoded in the mealybug genome (McCutcheon and von Dohlen, 2011; Husnik et al., 2013).
Here we investigated the symbiosis of the manna mealybug Trabutina mannipara (Ben-Dov, 1988), inhabiting the leaves and branches of the salt-secreting desert tree tamarisk (Tamarix nilotica; Waisel, 1961; Qvit-Raz et al., 2008; Finkel et al., 2011). Tremblaya cells in T. mannipara harbor an apparently novel gammaproteobacterial symbiont. Using a metagenomic approach to investigate the T. mannipara system and comparing our data with those available for P. citri, we asked whether the nested bacterial symbioses involving different intrabacterial symbionts in the two mealybug species display convergent patterns of evolution. Our results demonstrate that metabolic functions are partitioned in a highly complex yet surprisingly similar manner between the three symbiotic partners despite the polyphyletic origin of the intrabacterial symbionts in the two consortia. Moreover, bacterial genes involved in essential amino-acid and vitamin production are present in both mealybug genomes and were likely laterally acquired before T. mannipara and P. citri diversified.
Materials and methods
Sample collection and microscopy
Egg sacs of the mealybug T. mannipara were collected from tamarisk (T. nilotica; Waisel, 1961; Qvit-Raz et al., 2008; Finkel et al., 2011) in the Dead Sea area in Israel (31.271980°N, 35.215690°E). Mealybugs were transferred to Vienna while still attached to the tamarisk branches at ambient temperature. Egg sacs were stored at −80 °C for genomic DNA isolation.
We used fluorescence in situ hybridization, including an oligonucleotide probe specific for the 16S rRNA of the gammaproteobacterial symbiont of T. manniapara (Trabutinella-300: 5′-CAGTGTGGCTGTTTATCC-3′). Details on the fluorescence in situ hybridization procedure are given in Supplementary Information together with the light and transmission electron microscopic methods used for the visualization of the symbionts on semithin and ultrathin sections of the mealybug bacteriome, respectively.
Genome sequencing and assembly
Insects were dissected in 96% ethanol. Ventral sides of the mealybugs were opened and bacteriomes—usually showing up instantly as large and compact organs—were isolated by using fine forceps. Bacteriomes of 15–20 individuals were collected by pipetting, pooled, washed twice and homogenized in 1 × TE using a plastic pestle prior to DNA isolation. High molecular weight DNA was extracted by a CTAB-based isolation method (Zhou et al., 1996) using 1.5 v/v% polyvinylpyrrolidone in the extraction buffer.
Sequencing of a paired end library was performed on a HiSeq 2000 Illumina platform (Illumina Inc., San Diego, CA, USA). Nearly 177 million reads were obtained in both directions. Sequencing reads were filtered and trimmed by PRINSEQ (Schmieder and Edwards, 2011) and assembled with the SPAdes v3.1 genome assembler (Bankevich et al., 2012). In the initial assembly, using a subset of reads, three groups of contigs could be clearly separated by their G+C content and coverage. Assemblies were subsequently refined by recursive mapping of the reads and performing novel assemblies using the set of reads not mapped on non-target genomes, that is, contigs belonging to the insect host or one of the bacterial symbionts. Reads were mapped by the Burrows–Wheeler Alignment tool using the BWA-MEM algorithm (Li and Durbin, 2009). As a result, a single contig representing the circular chromosome of the gammaproteobacterial symbiont could be obtained with 1216-fold coverage. The genome of the outer symbiont Tremblaya is currently represented by four contigs with an average 592-fold coverage. Further bacterial contigs were not detected with reasonable coverage. Excluding those sequences that could be mapped back on the genomes of the bacterial symbionts, we generated a draft genome of T. mannipara with a coverage of ~11-fold. Only contigs >1000 bp were used for further analysis. Genome sequences have been submitted to the European Nucleotide Archive and are available under the accession numbers LT594522, FLRF01000001–FLRF01000004 and FKYK01000001–FKYK01037035.
Annotation, functional and comparative analysis of the symbiont genomes
The putative origin of replication was set with GenSkew (http://genskew.csb.univie.ac.at/). Synteny and rearrangements between genomes were visualized by Mauve (Darling et al., 2010).
We used an in-house pipeline, called ConsPred (Weinmaier et al., unpublished; available at https://sourceforge.net/p/conspred/) for gene prediction and annotation. Detailed description of the annotation method is available in the Supplementary Information. Genome annotations were manually curated using the UniPro UGENE tool (Okonechnikov et al., 2012). In order to identify potential pseudogenes, intergenic regions and hypothetical proteins were submitted to blastx searches against the non-redundant protein database (nr) of the National Center for Biotechnology Information and the UniProt Swiss-Prot databases. Protein domains were predicted according to the Conserved Domain Database (Marchler-Bauer et al., 2011) of National Center for Biotechnology Information. Pseudogenes were identified as remnants of genes, which are truncated or interrupted by internal stop codons and/or frameshifts showing high confidence (e-value<1e-3) blast hits to proteins with defined functions. Regions with premature stop codons were identified as pseudogenes if their length were <80% of the length of the reference sequences. Pseudogene coordinates were set according to the best blast hit against the Uniprot Swiss-Prot database.
Mobile genetic elements were checked by ISfinder (Siguier et al., 2006). Metabolic pathways were manually inspected using the Pathway Tools software (Karp et al., 2010) and the Ecocyc, Biocyc and Metacyc resources (Caspi et al., 2010). Clusters of orthologous groups shared by the relevant genomes or unique to the symbionts were identified by OrthoMCL using an 1e-5 e-value threshold (Li et al., 2003).
Phylogenetic placement of the symbionts
The relationship of the intrabacterial symbiont of T. mannipara with other symbionts of mealybugs was inferred by 16S rRNA-based phylogenetic analysis. A Bayesian tree was calculated with MrBayes v3.2.6 (Ronquist et al., 2012), using the GTR+I+G model with 4gamma categories. Two runs each with four chains were performed. The proportion of invariable sites and the gamma shape parameter were estimated from the data. Chains were stopped when convergence diagnostics fell <0.03; 50% majority consensus trees were generated with a relative burn-in of 25%. Tree calculations were performed on the CIPRES Science Gateway v.3.3. web interface (Miller et al., 2010).
A phylogenomic approach was also employed to characterize the relationship of the gammaproteobacterial symbiont of T. mannipara to other insect symbionts and free-living bacteria within the Enterobacteriaceae. For this, single copy marker genes were selected and aligned to the gammaproteobacterial reference data set integrated in Phyla-AMPHORA (Wang and Wu, 2013). Poorly aligned positions were excluded by Gblocks 0.91b (Castresana, 2000) using the default settings apart from the followings: minimum length of a block: 5, allowed gap positions: with half. A Bayesian phylogenetic tree was reconstructed from the concatenated alignment of 34 ribosomal proteins. We employed the CAT+GTR models (Lartillot and Philippe, 2004) as implemented in PhyloBayes MPI v1.6j (Lartillot et al., 2009, 2013) together with the Dayhoff 6 amino-acid recoding scheme, as suggested for phylogenetic reconstruction of symbiont lineages by Husník et al. (2011). Two chains were run until all discrepancies between the chains fell <0.1 and effective sample sizes were <100. The first 20% of the trees were discarded prior to generation of a majority consensus tree.
Phylogenetic trees were visualized with FigTree v1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/) and iTOL (Letunic and Bork, 2011).
Identification of contribution of the insect host to selected bacterial pathways
We investigated the role of the insect host in the T. mannipara symbiosis based on previous results by Husnik et al. (2013) on the symbiosis of the mealybug P. citri. Protein/DNA sequences already available for eukaryotic genes as well as genes of bacterial origin in the P. citri genome were used as queries in tblastn/blastn searches against the draft genome of T. mannipara, respectively. Genomic regions corresponding to the subject sequences of the best significant blast hits (e-value⩾1e-3) were extracted together with their flanking regions and were submitted for a blastx search against nr. Top hits (up to 100) were collected for each query and were used for phylogenetic analysis. Individual protein alignments were generated by the MAFFT L-INS-i algorithm (Katoh and Standley, 2013). Ambiguous positions were filtered with Gblocks as described above. Bayesian trees were generated in MrBayes v3.2.6 (Ronquist et al., 2012) as described for the 16S rRNA-based analysis above, except that we applied the WAG+I+G model with 4gamma categories.
Results and Discussion
T. mannipara contains a novel intrabacterial symbiont
The 16S rRNA of the betaproteobacterial symbiont of T. mannipara was most similar (97%) to ‘Ca. T. phenacola’ strains in other mealybugs, and its affiliation with the Tremblaya lineage was highly supported in the 16S rRNA-based phylogenetic analysis (data not shown). In contrast to the monophyletic origin of the primary symbionts of mealybugs, previous phylogenetic analyses of 16S and 23S rRNA genes suggested a polyphyletic origin of their secondary symbionts among the Enterobacteriaceae (Fukatsu and Nikoh, 2000; Thao et al., 2002). Consistent with this, our 16S rRNA-based analysis (Supplementary Figure S1) placed the inner symbiont of T. mannipara in a well-supported cluster with those of other mealybugs (Melanococcus albizziae, Australicoccus grevilleae, Amonostherium lichtensioides and Antonina pretiosa) from the tribe Trabutinini (Hardy et al., 2008) with which it shared 90.3–92.1% sequence similarity. This group clustered with other insect symbionts and the inner symbionts of other Trabutinini mealybugs (Paracoccus nothofagicola and Cyphonococcus alpinus) (Thao et al., 2002), although the exact relationships among these clades remain unresolved. Moranella endobia, the intrabacterial symbiont of P. citri, formed a relatively closely related yet distinct cluster together with the secondary symbiont of Planococcus ficus, in agreement with previous reports (Thao et al., 2002). Given its low degree of sequence similarity to known gammaproteobacterial species, the intrabacterial symbiont of T. mannipara represents a novel lineage within the Enterobacteriaceae. We thus propose to tentatively classify these symbionts as ‘Candidatus Trabutinella endobia’ (according to Murray and Stackebrandt, 1995); ‘Trabutinella’ alluding to the mealybug host, T. mannipara; ‘endo-bia’ (‘inside-living’) referring to the intrabacterial lifestyle of this bacterium).
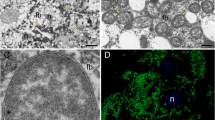
Fluorescence in situ hybridization using 16S rRNA-targeted-specific probes demonstrated that Trabutinella is present within the cells of Tremblaya, which in turn are located in the bacteriocytes of T. mannipara (Figure 1d). Light and electron microscopic observations on sections of T. mannipara bacteriomes showed that the intrabacterial Trabutinella are polymorphic and have a size of 2–7 μm in diameter (Figures 1a–c). Large (10–20 μm in diameter) cells of the outer bacterial symbiont Tremblaya contain up to 20 Trabutinella cells within their cytoplasm.

(a) Light microscopic micrograph at low magnification of a longitudinal section of the mealybug T. mannipara. The bacteriome (outlined in white) occupies a considerable part of the insect abdomen. (b) Light microscopic micrograph at higher magnification of the bacteriome. One of the bacteriocytes is outlined in white. The bacteriocytes present central nuclei with large nucleoli, indicating high cellular activity. Individual bacteriocytes contain up to seven enlarged bacterial symbionts (Tremblaya) in highly amorphous shapes, which themselves harbor several polymorphic bacteria (Trabutinella). (c) Transmission electron microscopic observation of a single host bacterium (Tremblaya) containing several bacteria (Trabutinella) within its cytoplasm. (d) Localization of ‘Candidatus Trabutinella endobia’ within ‘Ca. T. phenacola’ cells as seen with fluorescence in situ hybridization. A general bacterial probe (in green), a gammaproteobacterial probe (in red) and a Trabutinella-specific probe (in blue) were used; cells of Trabutinella appear white–purple, Tremblaya green.
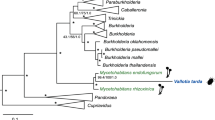
The genomes of bacterial symbionts are often characterized by skewed nucleotide composition and rapid sequence evolution, which hampers phylogenetic analysis and may result in grouping of unrelated taxa. Thus, in order to get further support for the relationship of Trabutinella and Moranella to other insect symbionts and free-living bacteria, we employed multilocus sequence data and advanced phylogenetic methods to overcome these biases, as suggested earlier (Husník et al., 2011). Phylogenetic analysis of a concatenated set of 34 ribosomal proteins (Figure 2, Supplementary Figure S2) showed that, although Trabutinella fell within the same monophyletic group as Moranella, they did not appear as sister taxa. This is consistent with our 16S rRNA-based analysis and confirms that the intrabacterial symbiosis in T. mannipara is a result of an infection of the Tremblaya cells independent from the one in P. citri. Trabutinella and Moranella were interleaved in a large group containing other facultative and obligate insect symbionts, such as Sodalis species, described as closest relative of Moranella (McCutcheon and von Dohlen, 2011), Baumannia in sharpshooters (Wu et al., 2006), Wigglesworthia in tsetse flies (Akman et al., 2002) and Blochmannia in carpenter ants (Gil et al., 2003; Degnan et al., 2005; Williams and Wernegreen, 2010). Although a common origin of this diverse set of symbionts is still debated, this group is consistent with one of the major symbiont lineages among the Enterobacteriaceae, which could be distinguished in recent comprehensive phylogenomic analyses (Husník et al., 2011; Manzano-Marín et al., 2015). Taken together, our data showed that phylogenetically different bacterial symbionts have independently infected Tremblaya during the evolution of T. mannipara and P. citri.

Phylobayes cladogram showing the affiliation of ‘Candidatus Trabutinella endobia’, intrabacterial symbiont of the mealybug T. mannipara, among Enterobacteriaceae. Insect symbiont lineages are colored in green. The complete phylogenetic tree with the original branch lengths is given in Supplementary Material (Supplementary Figure S2). The analysis is based on a concatenated set of 34 ribosomal proteins. Posterior probabilities are indicated on the internal nodes. Asterisks stand for posterior probabilities equal to 1. Nodes with a support of ⩽0.5 are collapsed.
A highly conserved Tremblaya genome and loss of functions in the intrabacterial symbiont from T. mannipara
We contrasted our results on the T. mannipara consortium to those reported for P. citri (McCutcheon and von Dohlen, 2011; Husnik et al., 2013; López-Madrigal et al., 2013). This analysis revealed that genomic sequences of Tremblaya were highly syntenic and harbored nearly identical sets of genes in the two systems in accordance with a monophyletic origin of the outer symbionts among mealybugs. Currently, three large contigs and a short fragment represent the genome of Tremblaya from T. mannipara. The latter corresponds to the 16S–23S rRNA and the 30S ribosomal protein S15 (rpsO) gene sequences, all duplicated in other Tremblaya strains. The draft genome of Tremblaya from T. mannipara (137 475 bp) was similar in size to the Tremblaya genome from P. citri (138 927 bp) (McCutcheon and von Dohlen, 2011). Only two genes were unique to Tremblaya from T. mannipara compared with Tremblaya from P. citri (McCutcheon and von Dohlen, 2011; Husnik et al., 2013; López-Madrigal et al., 2013). These encode an argininosuccinate lyase (argH), the final enzyme in the arginine synthesis, and a 16S rRNA methyltransferase (rsmH). Five genes (leuB, leuC, leuD, iscS and fdx (putative 2Fe-2S ferredoxin)) were present in the genome of Tremblaya from P. citri but were missing from the genome of the outer symbiont of T. mannipara. Among these, the leucine synthesis gene (leu) and the cysteine desulfurase gene (iscS) were pseudogenized and thus were likely recently inactivated in Tremblaya from T. mannipara. This is further supported by the observation that a functional homolog for each of these genes was found in the Trabutinella genome.
The genome of the intrabacterial symbiont Trabutinella showed only minimal synteny with that of Moranella from P. citri, which is consistent with the distinct evolutionary origin of these symbionts. Remarkably though, the Trabutinella genome was substantially more reduced having a size of 298 471 bp compared with the 538 294 bp genome of Moranella (McCutcheon and von Dohlen, 2011). The chromosome of Trabutinella had a G+C content of 32.6%: encoded for 237 proteins, one rRNA operon, and 41 tRNA genes. This means a >40% reduction in the number of protein-coding genes compared with Moranella. Moreover, the Trabutinella genome is likely still in the process of reduction as indicated by the presence of 27 pseudogenes (a feature typically not seen in tiny genomes of obligate symbionts) (Moran and Bennett, 2014).
Similarly to other obligate insect symbionts with small genomes (Moran et al., 2008; McCutcheon and Moran, 2012), most of the Trabutinella genome is devoted to replication, transcription, translation and to its symbiotic role, which—based on pathway reconstructions—is the complementation of Tremblaya with respect to essential amino-acid synthesis and vitamin provision of the insect host. In comparison to Moranella, >50% decrease in the percentage of genes involved in the central metabolism, coenzyme and lipid production, inorganic ion metabolism, transport mechanisms in general, cell envelope synthesis and cell division was observed (Supplementary Figure S3). For instance, while Moranella is still capable of glycolysis and can convert pyruvate to acetate and build up its own NAD+ pool (López-Madrigal et al., 2013), Trabutinella lacks all these capabilities except for the last two glycolytic enzymes (eno, pykF). Unlike Moranella, Trabutinella has lost the genes for the SUF machinery of the [Fe-S] cluster biosynthesis (only sufA is present). However, [Fe-S] clusters might be still synthetized by the combined action of both symbionts, with the help of the products of iscS (encoded in Trabutinella), iscU and erpA (in Tremblaya) (Vinella et al., 2009). Moreover, Trabutinella has apparently also lost its transporters and maintained only a putative mechanosensitive ion channel. This type of transporter, generally involved in the osmotic stress response, was also identified in Moranella among others. It has been proposed that mechanosensitive ion channels might facilitate the release of metabolites, perhaps even small proteins from Moranella, as skewed metabolic activity of the inner and outer symbionts might result in osmotic stress for the inner symbiont (López-Madrigal et al., 2013; López-Madrigal et al., 2013).
Surprisingly, Trabutinella does not encode a single known gene responsible for generation of the cell envelope, while Moranella still harbors most of the genes required for fatty acid and lipolysaccharide synthesis, some genes for peptidoglycan synthesis and two rod shape-determining proteins (McCutcheon and von Dohlen, 2011; López-Madrigal et al., 2013). As Tremblaya does not possess any of these functions either, synthesis of the cell envelopes of both bacterial symbionts likely fully relies on metabolites derived from the insect host in the T. mannipara system. A similar situation was described in mealybugs containing solely Tremblaya in psyllids, in whiteflies and in most members of the Auchenorrhyncha (Nakabachi et al., 2006; McCutcheon and Moran, 2010; Husnik et al., 2013; Jiang et al., 2013). However, how the process of insect host-mediated cell envelope synthesis is accomplished remains yet to be determined.
In accordance with the absence of cell wall synthesis genes, Trabutinella cells were polymorphic and appeared often in highly amorphous shapes (Figure 1). On the contrary, the cells of Moranella are typically rod shaped (von Dohlen et al., 2001), although apparently degenerative forms were also observed in mealybugs closely related to P. citri (Koga et al., 2013). It has been proposed that P. citri might complement missing functions in the peptidoglycan synthesis and degradation in Moranella owing to products of nine genes of bacterial origin present in the insect genome (Husnik et al., 2013). Differential expression of those genes might serve to regulate the integrity of Moranella cells and thereby the accessibility of Moranella products for the other symbiosis partners. In the absence of genes for peptidoglycan synthesis in both the bacterial symbionts and the T. mannipara draft genome, such a control mechanism seems unlikely to be at work in the T. mannipara symbiosis.
Bacterial genes in the T. mannipara genome
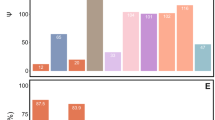
Out of the 22 genes of bacterial origin previously identified in the genome of the mealybug P. citri (Husnik et al., 2013), a set of nine genes was found in the draft genome of T. mannipara. Eight genes showed high similarity (>86% on the protein level) to their respective counterpart in P. citri, suggesting that their function is conserved and that they might be active in T. mannipara, as well. These genes are involved in essential amino-acid (cysK, dapF, lysA), biotin (bioA, bioB, bioD) and riboflavin (ribA, ribD) production (Figure 3). Function, putative origin and distribution of individual genes among the symbiosis partners in the T. mannipara versus P. citri consortia are shown in Figure 3 (genes of bacterial origin encoded in the insect genomes are highlighted by asterisks).

Convergent evolution of symbiotic roles in essential amino-acid synthesis, vitamin production and translation in T. mannipara (TM) and P. citri (PCIT). Colored boxes show the distribution of individual genes among the three symbiotic partners in the T. mannipara and P. citri consortia. Data for P. citri were taken from Husnik et al. (2013). Asterisks highlight genes of bacterial origin encoded in the T. mannipara and P. citri insect genomes. These genes were likely obtained by horizontal gene transfer during ancient associations of mealybugs with bacterial symbionts affiliated with the Alpharoteobacteria, Gammaproteobacteria or Bacteriodetes.
One gene, tms1 (tryptophan 2-monooxygenase oxidoreductase), shared only moderate similarity (amino-acid sequence identity=42.6%, similarity=60.4%) with its equivalent in P. citri, and its FAD-binding domain seemed to be disrupted. This gene was found in two copies in the P. citri genome, while only one was identified in T. mannipara. According to Husnik et al. (2013), tms1 was slightly overexpressed in the bacteriome. Its product is known to function in the bacterial synthesis of the phytohormone auxin, which is considered an important virulence factor in several plant-associated bacteria (Valls et al., 2006; Chen et al., 2007). The role of tms1 in mealybug symbioses remains unclear; however, the lower copy number and the disrupted functional domain found in T. mannipara suggest that this gene rather represents a non-functional relic of an ancient horizontal gene transfer event.
In addition, a ddlB (D-alanine-D-alanine ligase) pseudogene was found in the T. mannipara genome, which suggests that this bacterial gene has recently lost its function in this mealybug species. The ddlB pseudogene in T. mannipara contains frameshift mutations and internal stop codons, while ddlB was intact and highly active in the bacteriome of P. citri (Husnik et al., 2013). Degeneration of this gene, which is generally involved in peptidoglycan synthesis, is consistent with the absence of other genes accountable for bacterial cell envelope synthesis in the T. mannipara consortium.
Interestingly, all of the laterally acquired genes present in T. mannipara and P. citri and investigated here appeared as sister taxa in phylogenetic trees (Supplementary Figure S4). This indicates that they share a common origin and were present in ancestral mealybugs before P. citri and T. mannipara diversified. In accordance with previous findings (Husnik et al., 2013), our analyses suggested known symbionts of insects as potential donors, including Alphaproteobacteria (lysA from Ehrlichia, dapF from Rickettsia, ribD from Wolbachia) and Gammaproteobacteria (cysK from Sodalis, ribA from Arsenophonus). The affiliation of bioABD in T. mannipara could not be clearly determined, as they grouped both with Cardinium and members of the Rickettsiales (similarly to P. citri). Tms1 was putatively acquired from Pseudomanas, Burkholderia or Ralstonia species, microbes typically not found as symbionts of insects but encompassing auxin-producing plant-associated bacteria (Valls et al., 2006; Chen et al., 2007; Compant et al., 2008). Horizontal gene transfer from plant-associated bacteria to insects has recently also been proposed for the silkworm Bombyx mori (Li et al., 2011).
High degree of convergence between T. mannipara and P. citri symbioses
Symbiotic partners of the T. mannipara and P. citri systems partition the synthesis of essential amino acids in a highly similar manner (Figure 3). Although the majority of genes responsible for essential amino-acid synthesis are encoded in the Tremblaya genomes, only histidine might be produced by the outer symbionts alone in both consortia. The bacterial partners together can produce tryptophan and threonine, while synthesis of other essential amino acids likely requires additional gene products from the mealybug hosts.
In detail, although the majority of genes for histidine synthesis are present in all Tremblaya strains, the pathway is still incomplete in both symbiotic systems. Notably, Tremblaya of T. mannipara lacks exactly the same histidine-synthetizing genes as Tremblaya of P. citri and Tremblaya phenacola in P. avenae. Our observations thus further support that this pathway is indeed active but functions by a yet unknown mechanism in mealybug symbioses (Husnik et al., 2013).
The production of methionine, isoleucine, phenylalanine, arginine, valine and leucine likely requires products of eukaryotic genes (cystathionine beta (CBL)/cystathionine gamma (CGL), AAT, OAT, BCA), which were found in the T. mannipara genome, similarly to P. citri. These genes have also been identified in other insects, such as aphids (Hansen and Moran, 2011) and psyllids (Sloan et al., 2014), where they are typically overexpressed in the bacteriome compared with the rest of the insect body.
The synthesis of lysine and methionine seems to involve genes of bacterial origin present in both mealybug genomes (cysK, dapF, lysA). Similar to the synthesis of histidine, production of lysine is incomplete in both mealybug symbioses and might operate by a yet undetermined manner. The respective genes are also missing in members of Auchenorrhyncha (dapE) (McCutcheon and Moran, 2010) and in psyllids (argD) (Sloan et al., 2014), while the rest of the lysine synthesis pathway is encoded in the Sulcia and Carsonella symbionts, respectively.
It is striking that, in most of the essential amino-acid production pathways, exactly the same steps are carried out by the inner or the outer symbiont in T. mannipara and P. citri, despite the independent origin of the intrabacterial symbionts. For instance, intermediate genes in the chorismate synthesis are encoded in the inner symbionts, while the rest is present in Tremblaya in both systems (Figure 3). Moreover, the same genes are present exclusively in the inner symbiont in case of the synthesis of threonine and lysine. Altogether we found only five differences between the biosynthesis pathways for essential amino acids in the two consortia. (1) AroE in the chorismate (phenylalanine and tryptophan) synthesis is missing in the T. mannipara system, while it is present in P. citri. Lack of the shikimate dehydrogenase AroE despite the presence of all other genes in the chorismate synthesis has also been reported for psyllids, suggesting an alternate route which can bypass this gap in the pathway (Sloan et al., 2014). (2) The final enzyme of the arginine synthesis (argH) is encoded in Moranella in the P. citri consortium while it was identified in Tremblaya of T. mannipara. (3) Unlike in P. citri, dapA in the lysine synthesis does not exist in both symbionts but is only present in Tremblaya in T. mannipara. This is consistent with an increased reduction of functional redundancy in long-term symbioses. (4) Trabutinella carries out three steps of leucine synthesis (leuCDB), which are encoded by the Tremblaya genome in P. citri; and (5) cysE in the cysteine (and methionine) synthesis seems to be missing from both symbionts of T. mannipara, while it was found in Moranella in P. citri. As the bacterial cysteine synthase CysK encoded in the T. mannipara genome seems to be intact, it might contribute to the metabolism of cysteine if the necessary precursors were present. Sequences similar to CGL and CBL lyases were also found in the draft genome of T. mannipara, similar to P. citri, aphids and psyllids (Hansen and Moran, 2011; Husnik et al., 2013; Sloan et al., 2014). Thus cysteine might also be produced by the CGL activity from exogenous cystathionine, if that was available. Cystathionine, rather than cysteine might also be the substrate of the synthesis of methionine, as demonstrated by enzymatic assays for pea aphids (Russell et al., 2013). In this scenario, CBL activity of the host converts cystathionine into homocysteine, which can be converted to methionine by the homocysteine transmethylase MetE encoded in Tremblaya in case of both mealybugs. (Note that the insect host might also contribute to the production of methionine by the activity of a homocysteine S-methyltransferase, which was found in the genome of both P. citri (Husnik et al., 2013) and T. mannipara.). In addition, CGL might also have a role in the synthesis of isoleucine using homoserine, an alternative of cystathionine for 2-oxobutanoate production (Husnik et al., 2013; Russell et al., 2013).
In addition to essential amino acids, the cofactor riboflavin can also be produced in both mealybug symbiotic associations (Figure 3). Its synthesis is apparently performed in a similar manner in both consortia as it involves gene products from the gammaproteobacterial symbionts (ribEC) as well as insect host genes of bacterial origin (ribAD). The synthesis of biotin is carried out through Moranella and horizontally transferred genes in the P. citri genome, although several steps are missing from that pathway. Interestingly, symbionts of T. mannipara do not encode any of the genes involved in this pathway, although remnants of fabGZI are present as pseudogenes in the Trabutinella genome. Only the last three steps of the pathway might be carried out by bacterial genes encoded in the T. mannipara genome (bioADB), which could be used to build up biotin from 8-amino-7-oxononanoate, if that substrate would be available. This pathway is thus further reduced in the T. mannipara consortium compared with P. citri.
Similar to P. citri, the intrabacterial symbiont took over most of the translation-related functions in T. mannipara, including the ribosome-recycling factor, the peptide deformylase, the two release factors and all amino acyl tRNA synthetases. Only the three translation initiation factors (IF1–IF3) and two elongation factors (EF-Tu, EF-G) have been retained in Tremblaya as well as the inner symbiont in T. mannipara, exactly similar to P. citri (Figure 3). As translation-related genes were present in the genome of T. phenacola, which does not contain an intrabacterial symbiont, loss of these essential genes in Tremblaya of both T. mannipara and P. citri was likely facilitated by the acquisition of the inner symbionts (McCutcheon and von Dohlen, 2011; Husnik et al., 2013). However, our data imply that genes required for the initiation and elongation phase of translation are not subject of further reduction but instead are maintained in Tremblaya to perhaps provide some sort of independent control over protein production between the two symbionts.
Conclusions
In most plant sap-feeding insects, usually one of the bacterial symbionts is responsible for complete or nearly complete synthesis of individual metabolites. The high degree of metabolic complementarity, that is, the step by step division of labor between the nested symbionts in mealybugs is unusual, and is likely the consequence of their intimate intrabacterial association (McCutcheon and von Dohlen, 2011). Gradual degradation of the inner symbionts and release of their metabolites might facilitate the exchange of metabolites between the symbionts, thus allowing for a more complex partition of individual pathways between the partners in this association (McCutcheon and von Dohlen, 2011; Husnik et al., 2013; Koga et al., 2013). The highly congruent patterns of gene loss and metabolic complementarity observed in the T. mannipara and P. citri symbioses show that partition of the pathways is apparently not random as certain steps are carried out by the same symbiont in the two systems—despite the different evolutionary origin of the intrabacterial symbiont. A similar, yet far less complex situation has been observed among members of Auchenorrhyncha, where Sulcia synthesizes eight or seven essential amino acids while the remaining two or three are produced by different co-symbionts in different lineages, for instance. by Baumannia in sharpshooters, Hodgkinia in cicadas and Zinderia in spittlebugs (McCutcheon and Moran, 2010; Bennett and Moran, 2013).
A conceivable scenario explaining the observed similarities between the P. citri and T. mannipara symbioses would be that the Tremblaya ancestor was already infected by an (intra-)bacterial symbiont in ancestral mealybugs before P. citri and T. mannipara diverged. This ancient association would have facilitated reduction of the Tremblaya genome and has shaped its gene repertoire. We favor this scenario over alternative scenarios such as ancient co-obligatory or facultative associations of Tremblaya with another bacteriocyte-associated symbiont because of the high level of congruence between the loss of genes in different Tremblaya strains at intermediate steps of essential amino-acid synthesis pathways (such as in the synthesis of chorismate, lysine and threonine). These suggest that the primordial bacterial symbiont likely had an obligatory status and was already located within the cytoplasm of Tremblaya (facilitating exchange of metabolites and gene products) in the progenitor of the two mealybug species. The inner symbiont might have been subsequently replaced in the ancestor of P. citri and/or T. mannipara, with the new symbiont taking over the functions required by Tremblaya and at the same time allowing for loss of further genes from the Tremblaya genome, which would account for the observed differences between the two systems.
In summary, the identification of novel bacterial symbionts of Tremblaya in the mealybug T. mannipara and the genomic analysis of this consortium demonstrated an unexpected high degree of convergent evolution in these unique symbioses, in which—despite of the different evolutionary origin of one of the partners—both bacterial symbionts and the insect host act in concert in a highly complex manner. We also show that further reduction of essential functions in the already extremely deteriorated Tremblaya genome is possible.
References
Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M et al. (2002). Genome sequence of the endocellular obligate symbiont of tsetse flies Wigglesworthia glossinidia. Nat Genet 32: 402–407.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477.
Baumann L, Baumann P . (2005). Cospeciation between the primary endosymbionts of mealybugs and their hosts. Curr Microbiol 50: 84–87.
Baumann P . (2005). Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol 59: 155–189.
Ben-Dov Y . (1988). Manna scale, Trabutina mannipara (Hemprich & Ehrenberg) (Homoptera: Coccoidea: Pseudococcidae). Syst Entomol 13: 387–392.
Bennett GM, Moran NA . (2013). Small, smaller, smallest: The origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol Evol 5: 1675–1688.
Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F et al. (2010). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 38: D473–D479.
Castresana J . (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552.
Chen Z, Agnew JL, Cohen JD, He P, Shan L, Sheen J et al. (2007). Pseudomonas syringae type III effector AvrRpt2 alters Arabidopsis thaliana auxin physiology. Proc Natl Acad Sci 104: 20131–20136.
Compant S, Nowak J, Coenye T, Clément C, Ait Barka E . (2008). Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol Rev 32: 607–626.
Darling AE, Mau B, Perna NT . (2010). ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147.
Degnan PH, Lazarus AB, Wernegreen JJ . (2005). Genome sequence of Blochmannia pennsylvanicus indicates parallel evolutionary trends among bacterial mutualists of insects. Genome Res 15: 1023–1033.
Downie DA, Gullan PJ . (2005). Phylogenetic congruence of mealybugs and their primary endosymbionts. J Evol Biol 18: 315–324.
Finkel OM, Burch AY, Lindow SE, Post AF, Belkin S . (2011). Geographical location determines the population structure in phyllosphere microbial communities of a salt-excreting desert tree. Appl Environ Microbiol 77: 7647–7655.
Fukatsu T, Nikoh N . (2000). Endosymbiotic microbiota of the bamboo pseudococcid Antonina crawii (Insecta, Homoptera). Appl Environ Microbiol 66: 643–650.
Gil R, Silva FJ, Zientz E, Delmotte F, González-Candelas F, Latorre A et al. (2003). The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc Natl Acad Sci 100: 9388–9393.
Gruwell ME, Hardy NB, Gullan PJ, Dittmar K . (2010). Evolutionary relationships among primary endosymbionts of the mealybug subfamily Phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Appl Environ Microbiol 76: 7521–7525.
Hansen AK, Moran NA . (2011). Aphid genome expression reveals host–symbiont cooperation in the production of amino acids. Proc Natl Acad Sci 108: 2849–2854.
Hardy NB, Gullan PJ, Hodgson CJ . (2008). A subfamily-level classification of mealybugs (Hemiptera: Pseudococcidae) based on integrated molecular and morphological data. Syst Entomol 33: 51–71.
Husnik F, Nikoh N, Koga R, Ross L, Duncan RP, Fujie M et al. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153: 1567–1578.
Husnik F, McCutcheon JP . (2016). Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proc Natl Acad Sci USA 113: E5416–E5424.
Husník F, Chrudimský T, Hypša V . (2011). Multiple origins of endosymbiosis within the Enterobacteriaceae (γ-Proteobacteria): convergence of complex phylogenetic approaches. BMC Biol 9: 87.
Jiang Z-F, Xia F, Johnson KW, Brown CD, Bartom E, Tuteja JH et al. (2013). Comparison of the genome sequences of ‘Candidatus Portiera aleyrodidarum’ primary endosymbionts of the whitefly Bemisia tabaci B and Q biotypes. Appl Environ Microbiol 79: 1757–1759.
Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ et al. (2010). Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform 11: 40–79.
Katoh K, Standley DM . (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780.
Koga R, Nikoh N, Matsuura Y, Meng X-Y, Fukatsu T . (2013). Mealybugs with distinct endosymbiotic systems living on the same host plant. FEMS Microbiol Ecol 83: 93–100.
Lartillot N, Lepage T, Blanquart S . (2009). PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25: 2286–2288.
Lartillot N, Philippe H . (2004). A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21: 1095–1109.
Lartillot N, Rodrigue N, Stubbs D, Richer J . (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol 62: 611–615.
Letunic I, Bork P . (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res, W475–W478.
Li H, Durbin R . (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
Li L, Stoeckert CJ, Roos DS . (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189.
Li Z-W, Shen Y-H, Xiang Z-H, Zhang Z . (2011). Pathogen-origin horizontally transferred genes contribute to the evolution of Lepidopteran insects. BMC Evol Biol 11: 356.
López-Madrigal S, Balmand S, Latorre A, Heddi A, Moya A, Gil R . (2013). How does Tremblaya princeps get essential proteins from its nested partner Moranella endobia in the mealybug Planoccocus citri?. PLoS One 8: e77307.
López-Madrigal S, Latorre A, Porcar M, Moya A, Gil R . (2013). Mealybugs nested endosymbiosis: going into the ‘matryoshka’ system in Planococcus citri in depth. BMC Microbiol 13: 74.
Manzano-Marín A, Oceguera-Figueroa A, Latorre A, Jiménez-García LF, Moya A . (2015). Solving a bloody mess: B-vitamin independent metabolic convergence among gammaproteobacterial obligate endosymbionts from blood-feeding arthropods and the leech Haementeria officinalis. Genome Biol Evol 7: 2871–2884.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39: D225–D229.
McCutcheon JP, Moran NA . (2010). Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol 2: 708–718.
McCutcheon JP, Moran NA . (2012). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26.
McCutcheon JP, von Dohlen CD . (2011). An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr Biol 21: 1366–1372.
Miller MA, Pfeiffer W, Schwartz T . (2010), Creating the CIPRES Science Gateway for inference of large phylogenetic trees http://www.phylo.org/ accessed 9 April 2016).
Moran NA, Bennett GM . (2014). The tiniest tiny genomes. Annu Rev Microbiol 68: 195–215.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190.
Murray RGE, Stackebrandt E . (1995). Taxonomic note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int J Syst Bacteriol 45: 186–187.
Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA et al. (2006). The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314: 267.
Nikoh N, McCutcheon JP, Kudo T, Miyagishima S, Moran NA, Nakabachi A . (2010). Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS Genet 6: e1000827.
Okonechnikov K, Golosova O, Fursov M . (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28: 1166–1167.
Qvit-Raz N, Jurkevitch E, Belkin S . (2008). Drop-size soda lakes: transient microbial habitats on a salt-secreting desert tree. Genetics 178: 1615–1622.
Ronquist F, Teslenko M, Mark P, van der, Ayres DL, Darling A, Höhna S et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542.
Russell CW, Bouvaine S, Newell PD, Douglas AE . (2013). Shared metabolic pathways in a coevolved insect-bacterial symbiosis. Appl Environ Microbiol 79: 6117–6123.
Schmieder R, Edwards R . (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M . (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34: D32–D36.
Sloan DB, Nakabachi A, Richards S, Qu J, Murali SC, Gibbs RA et al. (2014). Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Biol Evol 31: 857–871.
Thao ML, Gullan PJ, Baumann P . (2002). Secondary (γ-Proteobacteria) endosymbionts infect the primary (β-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl Environ Microbiol 68: 3190–3197.
Valls M, Genin S, Boucher C . (2006). Integrated regulation of the type III secretion system and other virulence determinants in Ralstonia solanacearum. PLoS Pathog 2: e82.
Vinella D, Brochier-Armanet C, Loiseau L, Talla E, Barras F . (2009). Iron-sulfur (Fe/S) protein biogenesis: phylogenomic and genetic studies of A-type carriers. PLOS Genet 5: e1000497.
von Dohlen CD, Kohler S, Alsop ST, McManus WR . (2001). Mealybug β-proteobacterial endosymbionts contain γ-proteobacterial symbionts. Nature 412: 433–436.
Waisel Y . (1961). Ecological studies on Tamarix aphylla. (L.) Karst. Plant Soil 13: 356–364.
Wang Z, Wu M . (2013). A phylum-level bacterial phylogenetic marker database. Mol Biol Evol 30: 1258–1262.
Williams LE, Wernegreen JJ . (2010). Unprecedented loss of ammonia assimilation capability in a urease-encoding bacterial mutualist. BMC Genomics 11: 687.
Wu D, Daugherty SC, Van Aken SE, Pai GH, Watkins KL, Khouri H et al. (2006). Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol 4: e188.
Zhou J, Bruns MA, Tiedje JM . (1996). DNA recovery from soils of diverse composition. Appl Environ Microbiol 62: 316–322.
Acknowledgements
We thank Yair Ben-Dov for his help in identifying the sampling site and timing for collecting the mealybugs and Filip Husník for his guidance in the isolation of the bacteriomes. This study was funded by the Austrian Science Fund (FWF) project P22533-B17. Work in the Belkin laboratory (OMF and SB) was supported by United States–Israel Binational Science Foundation grant 2010262.
Note
A paper by Husnik and McCutcheon (Husnik and McCutcheon, 2016) that appeared during the revision/proof stage of this article demonstrated that symbiont replacement also occurred in other mealybug lineages.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Szabó, G., Schulz, F., Toenshoff, E. et al. Convergent patterns in the evolution of mealybug symbioses involving different intrabacterial symbionts. ISME J 11, 715–726 (2017). https://doi.org/10.1038/ismej.2016.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.148
This article is cited by
-
PacBio Hi-Fi genome assembly of Sipha maydis, a model for the study of multipartite mutualism in insects
Scientific Data (2024)
-
Stereo-microscopic study of the anatomy of papaya mealybug Paracoccus marginatus (Williams and Granara De Willink)
International Journal of Tropical Insect Science (2024)
-
The transcriptome of Icerya aegyptiaca (Hemiptera: Monophlebidae) and comparison with neococcoids reveal genetic clues of evolution in the scale insects
BMC Genomics (2023)
-
Symbioses shape feeding niches and diversification across insects
Nature Ecology & Evolution (2023)
-
Molecular characterization, ultrastructure, and transovarial transmission of Tremblaya phenacola in six mealybugs of the Phenacoccinae subfamily (Insecta, Hemiptera, Coccomorpha)
Protoplasma (2019)
