
Abstract
Whether a small cell, a small genome or a minimal set of chemical reactions with self-replicating properties, simplicity is beguiling. As Leonardo da Vinci reportedly said, ‘simplicity is the ultimate sophistication’. Two diverging views of simplicity have emerged in accounts of symbiotic and commensal bacteria and cosmopolitan free-living bacteria with small genomes. The small genomes of obligate insect endosymbionts have been attributed to genetic drift caused by small effective population sizes (Ne). In contrast, streamlining theory attributes small cells and genomes to selection for efficient use of nutrients in populations where Ne is large and nutrients limit growth. Regardless of the cause of genome reduction, lost coding potential eventually dictates loss of function. Consequences of reductive evolution in streamlined organisms include atypical patterns of prototrophy and the absence of common regulatory systems, which have been linked to difficulty in culturing these cells. Recent evidence from metagenomics suggests that streamlining is commonplace, may broadly explain the phenomenon of the uncultured microbial majority, and might also explain the highly interdependent (connected) behavior of many microbial ecosystems. Streamlining theory is belied by the observation that many successful bacteria are large cells with complex genomes. To fully appreciate streamlining, we must look to the life histories and adaptive strategies of cells, which impose minimum requirements for complexity that vary with niche.
Similar content being viewed by others

Editor's note
Professor Stephen Giovannoni was invited to contribute a Winogradsky review based on his recognized contributions to microbial ecology of the oceans, including discovery, isolation and detailed analyses of the globally abundant, but previously uncharacterized SAR11 clade, that he named Pelagibacter. His studies have revealed remarkable insights into the genomics and metabolism of Pelagibacter and have made major contributions towards understanding the ecological role of bacteria in marine ecosystems. Insights gained from his studies have increased understanding of streamlined metabolism and its impact on ocean ecology, as exemplified in an associated paper in this issue for thiamin (vitamin B1) biosynthesis.
Introduction
Although the salient feature of streamlined cells is their small genome size, ‘streamlining’ refers more generally to selection that favors minimization of cell size and complexity. In theory, streamlining will not matter much in nutrient-rich environments, but it can be critical to success in nutrient-poor environments, where either gathering a larger share of nutrient resources, or using them more efficiently, can increase success. Here, we define ‘success’ in its neo-Darwinian sense––high gene frequencies––because in microbial ecology today this is the most common measurement of abundance and diversity. Indeed, this review mainly addresses genome streamlining, because genomic data are so much more accessible, but readers should keep in mind that cell size reduction can be a result of the same selective pressure, with smaller cells in principle benefitting not just by reduced replication costs, but also by higher surface-to-volume ratios that confer superior nutrient transport properties (Button, 1991; Sowell et al., 2008; Moya et al., 2009).
The idea that small, simple, compact cells can be more efficient is beguiling, and found expression in early discourse about the nature of microbial evolution (Doolittle and Sapienza, 1980; Orgel and Crick, 1980). However, streamlining is a modern theory that fits into a spectrum of ideas that have historically been used to discuss microbial adaptations to life in extremely dilute, natural environments where success is determined by resource competition. Below, we discuss this background, and also examine genome streamlining in the context of new ideas that explore the potential implications of streamlining for co-evolutionary dynamics in microbial communities (Morris et al., 2012). We also dwell on the example of Pelagibacter to illustrate how non-canonical metabolic pathways can evolve by streamlining, altering our perspectives of metabolic adaptation and gene regulation (Smith et al., 2010, 2013).
Evidence is emerging that streamlined genomes are much more common than was previously thought. Ironically, most of the data supporting streamlining theory came from the study of cultured cells, in particular, a handful of organisms that attracted attention because culture-independent studies demonstrated that they were highly successful in nature. But now, with single-cell genomics a practical reality and better metagenomic assemblies, evidence is accumulating that small genomes are common, and may partly explain why many organisms that are successful in nature are also challenging to culture.
Microbiological concepts for understanding growth at low-nutrient extremes
Some well-established concepts in microbiology provide an important framework for this review. Oligotroph was coined in reference to metabolism and growth at very low-nutrient concentrations that are common in nature, whereas copiotroph, a reference to growth at high-nutrient concentrations, has become almost a synonym for common chemoheterotrophic bacteria that grow easily and rapidly in culture (Fry, 1990; Lauro et al., 2009). Some organisms (probably many) can do both, and are called facultative oligotrophs. The terms r- and k-strategist are sometimes used interchangeably with copiotroph and oligotroph, but refer to success by replicating rapidly when conditions are favorable, versus slow, steady growth (Klappenbach et al., 2000). There is no a priori reason why oligotrophs cannot be r-strategists or copiotrophs k-strategists; whereas the terms copiotroph and oligotroph refer to cellular interactions with nutrients, defensive measures to evade predation by viruses and protists can have a large role in determining whether an organism is an r or k-strategist. From our experience with oligotrophic ocean systems, the most successful microorganisms are often obligately oligotrophic k-strategists that successfully compete for dissolved, labile organic matter, whereas the same systems harbor rare, facultatively oligotrophic r-strategists that respond successfully to relatively rare opportunities, and probably have complex life histories (Vergin et al., 2013).
Don Button, who pioneered the development of methods for isolating bacterial cultures in media made from natural seawater and lake water, also developed a theory that defined oligotrophs in terms of uptake kinetics, membrane transporter properties, metabolic pathway properties and the surface-to-volume ratios of cells (Button, 1991, 1998). His theory directly addressed the varying abilities of cells to effectively compete at very-low-nutrient concentrations. A fundamental tenet of his kinetic theory was that successful oligotrophic bacteria are examples of process optimization; they must be large enough to house the required genome and processes, while minimizing size and complexity to make the most efficient use of resources. Interestingly, evaluation of Button’s concepts from the post-genomic perspective has not been straightforward because many of the cellular features that were important in his mathematical models––for example, surface-to-volume ratios, per-cell protein numbers, and enzyme substrate specificity and kinetic properties––are not readily predictable from genome sequence.
Post-genomic efforts have attempted to define oligotrophs and distinguish them from copiotrophs in terms of genome properties. For example, Lauro et al. (2009) and others used clustering methods based on self-organizing maps to identify genome features associated with fast and slow growing lifestyles. In one of the few examples that attempted to link genome features to experimental measurements of function, Dethlefsen and Schmidt (2007) compared the translational properties of fast and slow growing bacteria, and concluded that rate of protein synthesis normalized to the mass of the protein synthesis system is three- to fourfold higher among bacteria that respond rapidly to nutrient availability.
Streamlining theory
The first very small genomes observed in free-living cells were reported in cyanobacteria of the genus Prochlorococcus (1.66–2.41 Mb; Rocap et al., 2003; Dufresne et al., 2005), chemoheterotrophic alphaproteobacteria in the order Pelagibacterales (SAR11; 1.28–1.46 Mb; Giovannoni et al., 2005; Grote et al., 2012) and obligate methylotrophs of the OM43 clade (1.30 Mb; Giovannoni et al., 2008; Figure 1). Importantly, the genomes from these free-living organisms, although small, had very different characteristics than that observed in obligate symbiotic, parasitic or commensal (SPC) microorganisms. Streamlined genomes are typically characterized by: (1) small genomes with highly conserved core genomes and few pseudogenes; (2) low ratios of intergenic spacer DNA to coding DNA; (3) low numbers of paralogs (Giovannoni et al., 2005; Wrighton et al., 2012; Sabath et al., 2013; Swan et al., 2013). Streamlining theory emerged to explain these observations. The essence of streamlining theory is that selection is most efficient in organisms that have large effective population sizes, and favors cell architecture that minimizes resources required for replication (Lynch and Conery, 2003; Dufresne et al., 2005; Giovannoni et al., 2005; Lynch, 2006; Giovannoni et al., 2008; Dupont et al., 2011; Grzymski and Dussaq, 2012).

Alternate pathways of genome reduction. The mechanisms leading to small genomes in symbiotic and commensal organisms (for example, mycoplasmas) are fundamentally different from the process that lead to the small genomes that have recently been discovered in many free-living organisms from natural systems.
Alternative theories that could explain small-genome size in free-living microbial populations, for example, gene loss by genetic drift or mechanisms isolating genomes from horizontal gene transfer, conflict with reports of strong purifying selection and frequent horizontal transfer of genetic material in streamlined microorganisms (Kettler et al., 2007; Vergin et al., 2007; Wilhelm et al., 2007).
Streamlining theory is based on the idea that under some circumstances selection can favor the minimization of cell complexity. Lynch and Conery (2003) supplied important background when they argued the opposite––that, in populations with low Ne, ‘selfish’ DNA could propagate because selection was too inefficient to eliminate it. Lynch and Conery based their ideas on theory developed by Kimura (1968), who showed that genetic drift occurs when |s| <1/(2 Ne), where s is the selective advantage, that is, change in fitness, and Ne is the effective population size. To explain small microbial genomes, streamlining theory applied the converse argument: in populations with large Ne values, selection will be very efficient (Giovannoni et al., 2005). Ne is usually measured indirectly, by measuring θ values (θ=2Neμ in haploid organisms, where μ is the mutation rate per locus per generation). Estimates of Ne obtained by this method are typically low, compared with the census size of populations obtained by counting cells. A number of explanations have been put forward to account for this (Fraser et al., 2009), and it remains a stumbling block to streamlining theory that measured Ne values are often too low.
Genomes belonging to SPC organisms, growing in close association with other cells, can have much smaller microbial genomes than those of streamlined, free-living cells. In many cases, these associations are between bacteria and eukaryotes, although there are examples of bacterial and archaeal symbionts with other bacteria or archaea (Baker et al., 2010). Genome reduction in SPC organisms occurs by evolutionary processes that are distinctly different from those that define streamlining evolution, and leaves different genomic signatures (McCutcheon and Moran, 2012). These two pathways of genome reduction are contrasted in Figure 1. Small genomes of the SPC type have been attributed to genetic drift that produces very different genomic signatures, including the expansion of noncoding genetic material, elevated rates of non-synonymous substitution and loss of biosynthetic pathways, resulting in increased prototrophy (Kuo et al., 2009). To illustrate the SPC pathway of genome reduction, we offer a hypothetical example. Consider a bacterium entering an obligate symbiosis. If the bacterium is constantly in association with its host, some of its biosynthetic operons may be turned off permanently by transcriptional control mechanisms because products of these operons, for example, amino acids, are produced by the host. Unexpressed, these biosynthetic genes are no longer under selection to provide the function they evolved for, and will decay to pseudogenes by mutation. In this scenario, the eventual loss of pseudogenes is ensured by a proposed deletional bias, which favors the random loss of DNA from bacterial genomes during replication (Mira et al., 2001). Alternatively, even non-essential genes with value can be lost by genetic drift, which is particularly likely in populations that have a low Ne––a category that includes many symbionts. Perhaps the best examples of this are Buchnera spp., which are bacterial symbionts of aphids (van Ham et al., 2003).
In the discussion above, we emphasize that increases in the discriminating power of selection can favor streamlining, leaving open the question of how small genome size confers a fitness advantage. In streamlined marine organisms there is a trend to higher proportions of nucleotides (A+T) and amino acids (for example, the substitution of lysine for arginine) that have lower N content, indicating that streamlining selection acts to reduce the amount of nitrogen needed for cell replication (Giovannoni et al., 2005; Grzymski and Dussaq, 2012). Evidence that streamlining selection is driven by fitness advantages of reduced cellular P quotas is, so far, not convincing, but that may be because the data needed to test this hypothesis are less readily available (Vieira-Silva et al., 2010); in the oceans, N and P tend to be co-limiting (Falkowski et al., 1998). Comparative studies of streamlining in P-limited and P-replete systems might shed light on this issue.
Reductions of cellular N and P quotas are not the only factors that have potential to drive streamlining selection. Streamlined marine bacteria include some of the world’s smallest organisms (Chisholm et al., 1992; Rappé et al., 2002; Ghai et al., 2013), raising the possibility that increased surface-to-volume ratios might also drive streamlining in some cases. A metaproteomics study of Pelagibacter in the ultra-oligotrophic Sargasso Sea showed that a strikingly high (67%) proportion of cellular protein is devoted to transport functions, suggesting that high surface-to-volume ratios are a factor in the success of these very small (ca 0.01 μm3), streamlined cells (Sowell et al., 2008). In a twist on this perspective, Thingstad argued that bacterioplankton may increase their cell size to succeed by defense specialism––growing too large for their predators. He theorized that the surface-to-volume ratio is less important than the surface-to-‘cell requirements of a limiting element’, and that larger cells can maintain the benefits of smaller cells by scaling up their cell composition by accumulating non-limiting nutrients such as carbon in an N- and P-limited system (Thingstad et al., 2005). The theory that selection favors small genomes because they can be replicated more rapidly, thus shortening generation times, has largely been ruled out by studies that found no correlation between generation time and genome size (Mira et al., 2001; Lynch, 2006; Touchon and Rocha, 2007; Vieira-Silva and Rocha, 2010).
New data show streamlining might be more prevalent than once thought
Recently, reports of small genomes in uncultured taxa have flooded into the field, providing strong support for the idea that the prevalence of streamlined bacteria in nature has been underestimated because they are challenging to culture. Early indications that streamlining might be a pervasive phenomenon came from flow cytometry showing that, whereas most cultivated oligotrophic isolates have genome sizes of >2.9 Mb, natural microbial plankton communities have genome sizes much lower, in the range of 1–2 Mb (Button and Robertson, 2001). Later, metagenomic data yielded effective genome sizes of 4.7 Mb for soil bacteria, and 1.6 Mb for marine bacteria from the Sargasso Sea, a highly oligotrophic site (Raes et al., 2007). Since that early work was published, many estimates of average microbial genome size from metagenomic data have been reported (Figure 2; Frank and Sorensen, 2011; Oh et al., 2011; Quaiser et al., 2011; Xia et al., 2011; Eiler et al., 2013). Estimates of average genome size from metagenomic data have now been reported for many systems, including fresh water, brackish water and aquifers (Figure 2).

Average microbial genome sizes estimated from metagenomic data, adapted from Angly et al. (2009), with additional data from Frank and Sorensen (2011), Oh et al. (2011), Quaiser et al. (2011), Xia et al. (2011), Eiler et al. (2013).
Dupont et al. (2011) estimated the genome size of the uncultured, highly abundant marine gammaproteobacterium SAR86 at 1.25–1.7 Mb, whereas Ghai et al. (2013) measured the cell volume of SAR432 marine Actinobacteria at ca 0.013 mm3, and used fosmids to estimate its genome size at <1 Mb. Swan et al. (2013) analyzed 41 single amplified genomes (SAGs) from surface populations of marine plankton, and concluded that many abundant marine bacteria exhibit the same characteristics of streamlining seen in Pelagibacter, Prochlorococcus and OM43. They also observed that, within taxonomic groups containing both SAGs and cultured representatives, there are differences between SAGs and their cultured relatives in genomic signatures of streamlining (for example, Rhodobacteraceae in Figure 4). The new evidence of small genomes is not limited to the oceans. Wrighton et al. (2012) assembled the genomes of four candidate divisions of uncultured bacteria from aquifer metagenomic data and estimated their genome sizes at <2 Mb. Following up, the same research team reconstructed additional genomes of four candidate phyla (SR1, WWE3, TM7 and OD1) from aquifer sediment and found very small genomes (0.7–1.2 Mb) that lacked identifiable biosynthetic pathways for several key metabolites (Kantor et al., 2013).
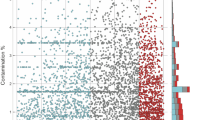
Figure 3 shows the % noncoding (spacer) DNA versus genome length for all published bacterial genomes in the IMG v400 database (see Supplementary Methods), as well as those recovered from marine surface water by single-cell genome amplification (Swan et al., 2013). The genomes of cultured bacteria that are known to be highly abundant in nature, and genomes recovered without cultivation are smaller than average. Previous analysis of cultured bacterial and archaeal genomes showed that size tends to be bimodal, with peaks at ca 2 and 5 Mb (Koonin and Wolf, 2008). Although the distribution of genome sizes among genomes with a status of ‘Finished’ in IMG v400 is not significantly different from unimodality (see Supplementary Methods), including all ‘Finished’, ‘Permanent Draft’ and ‘Draft’ genomes supports the trend observed by Koonin and Wolf (2008); (Hartigan’s Dip Test (Hartigan and Hartigan, 1985), N=5689, D=0.01, P=7.56 × 10−5), with separation into two populations at ∼3.7 Mbp (Figure 3). Such results pouring in from both continued exploration of whole genomes with single-cell genomics and metagenomic assembly indicate that small genomes (for example, 0.7–1.6 Mb) are common in nature. However, natural ecosystems remain vastly under-sampled with technologies that resolve genome size, and thus the true scope of streamlining is still unknown.

The % of noncoding (spacer) DNA versus genome length for all published bacterial genomes in IMG v400 and estimated genome size for single-amplified genomes (SAGs) from Swan et al. (2013). Streamlined (SAR11, Prochlorococcus, symbionts) and non-streamlined important marine taxa (Vibrionaceae, Rhodobacteraceae, Alteromonadaceae) have been highlighted (legend). Organisms where streamlining is driven by nutrient-limitation, such as SAR11 and those represented by the uncultured single-amplified genomes from Swan et al. (2013), have low % noncoding DNA and tend to fall at the extremes of the distribution of residuals from the linear regression line (black line). Conversely, organisms where streamlining is driven by symbiosis tend to maintain a broad range of % noncoding DNA, despite their small genomes. Histograms (top, right) show the distribution of points across each axis. The distribution of genome lengths (top) was significantly different to a unimodal distribution (Hartigan’s Dip Test; Hartigan and Hartigan, 1985), N=5689, D=0.01, P=7.56 × 10−5) with a separation of the two modes at ∼3.7 Mbp.
Life history strategy is also a factor in genome size
One of the biggest challenges to streamlining theory is explaining why many successful, abundant microorganisms, including some pelagic marine bacteria, do not have small genomes. The solution to this paradox lies in understanding that evolution toward small cell size and genome simplicity is not the only successful strategy in nature. The limit of genome reduction as a function of streamlining must be balanced with the need to maintain sufficient functional complexity to succeed within a niche. Thus, the many genomic signatures of streamlining, such as low GC, low percentage of noncoding nucleotides and few pseudogenes, should be observable across a range of genome sizes. Evidence of this is seen in the many points falling below the regression line in Figure 3. If this model is correct, then those free-living organisms with the smallest genomes (for example, Prochlorococcus, Pelagibacterales and OM43) occupy niches that require minimal functional complexity, and have been further reduced in genome complexity by selection acting to minimize the resources needed to replicate. Thus, strategies for resource competition may be as important as streamlining selection is for determining genome size.
In bacteria, increasing functional complexity typically implies regulation in response to environmental change, for example, most model chemoheterotrophic microorganisms turn on and off operons for the catabolism of a wide range of compounds, or shift their metabolism completely. In general, the proportion of microbial genomes devoted to regulation decreases with size, as an increasingly large proportion of the genome is devoted to core functions (Ranea et al., 2005). Thus, in principle, small genomes are likely to be found in organisms that occupy fundamental niches that are relatively invariant. Conversely, organisms that transition between aerobic and anaerobic environments (Manganelli et al., 1999), surface attachment (Ishimoto and Lory, 1989) and circadian rhythms (Yoshimura et al., 2007) require major shifts in metabolism and/or physiology. Global regulation of these processes is commonly encoded by σ-factors.
In the discussion that follows we use sigma factors provide a proxy for life history complexity. σ-Factors are polypeptides that combine with DNA-dependent RNA polymerase (RNAP) to form an RNAP holoenzyme capable of transcribing the DNA template. The role of the σ-factor in the holoenzyme is twofold: (1) RNAP is unable to initiate transcription without the σ-factor. (2) The σ-subunit of the RNAP holoenzyme can recognize specific promoter sequences and can thus initiate transcription of specific genes, orchestrating a metabolic and/or physiological response to changing environments (reviewed in Wösten, 1998). Housekeeping σ-factors, such as σ70 in Escherichia coli (encoded by rpoD), are responsible for the transcription of most genes expressed in exponentially growing cells and it is thought that at least one copy of a σ70-like homolog can be found in almost all bacterial genomes. Other non-essential σ-factors include stationary phase σ-factors (RpoS); flagellar σ-factors (σ28, WhiG); heat-shock σ-factors (σ32, SigB/C); sporulation σ-factors; nitrogen utilization σ-factors (σ54) and a broad range of extracytoplasmic function σ-factors controlling expression of, among other things, alginate biosynthesis, iron uptake, antibiotic production and virulence factors (Wösten, 1998).
It is reasonable to assume that the more complex the life history of an organism, the greater the number of σ-factors that will be encoded in its genome. Figure 4 shows how the number of σ-factor homologs in a genome varies across genome size for all published bacterial genomes in the IMG v400 database (see Supplementary Methods), along with the streamlined genomes of uncultured marine SAGs from Swan et al. (2013). The distribution of σ-factors shown in Figure 4 highlights two interesting observations. First, unlike streamlined organisms, intracellular parasites and symbionts tend to encode very few σ-factors while maintaining a broad range of % noncoding DNA (Figures 3 and 4). This is a marked difference resulting from the alternate genome reduction pathways outlined in Figure 1. By using the number of accumulated mutations in pseudogenes as a proxy for the length of time since a gene transitioned to a pseudogene, Babu (2003) showed that the high number of pseudogenes in Mycobacterium leprae (1116 compared with 6 in the close relative M. tuberculosis) was likely the result of an initial loss of σ-factors through genetic drift. This was followed by a cascade of pseudogene formation as genes under σ-factor regulation fell under relaxed selection pressure. Copiotrophic taxa such as Vibrionaceae, Bacteroidetes and Rhodobacteraceae tend to have σ-factor distributions that span the regression line (Figure 4), whereas streamlined genomes such as SAR11, Prochlorococcus, OM43 and uncultured SAGs tend to fall well below it, with SAR11 located at the extreme edge of the distribution, lacking even RpoS (Giovannoni et al., 2005; Lauro et al., 2009). Interesting exceptions to this rule include Prochlorococcus strains MIT9303 and MIT9313, which contain almost twice as many σ-factor homologs as other Prochlorococcus strains, as well as higher % GC content (50.01% and 50.74%, respectively, compared with 30–35% GC content for other Prochlorococcus strains). These genomes are also significantly larger than other Prochlorococcus strains (2.68 and 2.41 Mb, respectively, compared with approximately 1.6–1.8 Mb, respectively, for other Prochlorococcus strains), and are the earliest clade to diverge from their non-streamlined Synechococcus ancestor (Rocap et al., 2002; Kettler et al., 2007).

The number of σ-factor homologs versus genome length for published bacterial genomes in IMG v400 (grey), up to 5 Mbp in length with important marine microbial taxa highlighted (legend). Fitting the data across all genome lengths (inset) with a Poisson distribution showed evidence of overdispersion (Φ=5.50) therefore regression was performed using a negative binomial generalized linear model with a log-link (green line main figure, black line inset. Lighter green ribbon represents 95% confidence intervals of the model). The explained deviance of the negative binomial model was 0.73. Uncultured SAG representatives from Swan et al. (2013), SAR11, Prochlorococcus and symbionts fell below the regression line, supporting the hypothesis that low numbers of σ-factors are a consistent feature of streamlined genomes. SAGs from Verrucomicrobia fell above and below the regression line, supporting the findings of Swan et al. (2013) that these genomes can be found in both streamlined and non-streamlined varieties.
Second, many taxa have fewer σ-factors and % noncoding DNA (Figures 3 and 4) than would be expected from their genome size, even when the genome size is relatively large, demonstrating that streamlining is not synonymous with small genomes. It is postulated that such organisms are likely to have a relatively simple life history, albeit in a niche which requires complex pathways for the assimilation of available resources. SAGs from uncultured Rhodobacteraceae contained fewer σ-factors than their cultured counterparts, perhaps suggesting an adaptation to a less dynamic microbial niche (Swan et al., 2013).
The loss of complex regulatory mechanisms such as those controlled by σ-factors can be contrasted with regulation requiring a minimum of biochemical machinery in the form of small or noncoding RNAs, including riboswitches. Riboswitches are cis-acting noncoding regulatory RNAs that bind small molecules or respond to temperature to either promote or repress translation of a (usually) downstream transcript (Dambach and Winkler, 2009). Other regulatory noncoding RNAs can be trans-acting and interact with proteins and riboswitches (Waters and Storz, 2009). Although not unique to organisms with streamlined genomes (Winkler and Breaker, 2005), noncoding RNAs represent a simple solution to regulation in the context of fewer genes and many have been cataloged in Pelagibacter (Meyer et al., 2009; Tripp et al., 2009; Smith et al., 2010) and Prochlorococcus (Steglich et al., 2008). Prochlorococcus MED4 contains only 6 identified protein regulators compared with the 32 found in E. coli, and few σ-factors (Figure 4). However, its genome includes 24 noncoding RNAs, which were upregulated under light- and phage-induced stress (Steglich et al., 2008). Similarly, Pelagibacter lacks common genes for glycine biosynthesis, and therefore has a conditional requirement for glycine or glycine precursors (for example, serine, glycine betaine or glycolic acid; Tripp et al., 2009). However, this ostensibly deleterious deletion is compensated for by a glycine riboswitch upstream of the malate synthase that has been postulated to regulate central metabolism based on internal glycine levels (Tripp et al., 2009).
Living streamlined
In the section above, we emphasize general relationships between metabolic complexity, life cycle complexity and regulation, which dictate that, at the small end of the genome size spectrum, cellular activity is simple and efficient. But, each streamlined organism studied so far has unusual characteristics that appear to be dictated by the dispensable functions of the niche. All Prochlorococcus genomes lack some DNA repair genes, two genes (psbU/V) involved in the stabilization of the PS II oxygen-evolving complex, three genes encoding glycolate oxidase, a complex involved in photorespiration, and genes encoding two ABC transporters that are thought to be used for the uptake of sucrose (Partensky and Garczarek, 2010). Prochlorococcus also lack the katG gene that encodes a catalase/peroxidase, and therefore are sensitive to hydrogen peroxide, although they can benefit from the presence of katG+ cells in the neighborhood (Morris et al., 2011). The leaky function of KatG has been used as an example to illustrate the concepts of the Black Queen Hypothesis (BQH; Morris et al., 2012). One interpretation is that Prochlorococcus, and other katG− genotypes, are social cheaters that save themselves from the cost of synthesizing KatG by relying on other community members to detoxify hydrogen peroxide.
The deep-branching lineage of marine Actinobacteria that was described recently by Ghai et al. (2013) is estimated to have a genome of <1 Mb, a very low GC content (33%) and a cell size about the same as Pelagibacter (ca 0.013 μm). Interestingly, actinobacterial-specific genes for mycothiol biosynthesis and coenzyme F420-dependent enzymes were found in the genome, but, not surprisingly, there was little mention of missing functions––the identification of gene losses and reconstruction of metabolic pathways re-arranged by streamlining selection often can take years of study.
The genome of the methylotrophic betaproteobacterium OM43 is strikingly small compared with other methylotrophs, but the most notable genes absent were mxaF and mxaI, which code for large and small subunits of methanol dehydrogenase. Instead, OM43 relies on an alternative methanol dehydrogenase, xoxF, which only recently was recognized as a bona fide methanol dehydrogenase alternative. OM43 is a coastal species that is found associated with phytoplankton (Morris et al., 2006). Recent reports of high turnover rates for methanol in the ocean surface (Dixon et al., 2011), a report describing many alternative OM43 substrates (trimethylamine, trimethylamine oxide, dimethylamine and methyl chloride; Halsey et al., 2012), and a report that OM43 xoxF was 2.3% of the identified spectra in a productive coastal ocean metaproteome (Sowell et al., 2011), all suggest future studies will aim to understand this unusual organism.
The studies from Wrighton et al. (2012) and Kantor et al. (2013), referred to above, differed markedly from others that reported streamlining, in that they focused on a freshwater aquifer that had been amended with acetate. Nonetheless, these studies reported multiple, very small genomes from candidate phyla, all of them lacking respiration and instead apparently relying on fermentation for energy. In most of the genomes, complete biosynthetic pathways for nucleotides, lipids and many amino acids could not be identified, leading the authors to conclude that these organisms might be auxotrophic for many essential metabolites. In this anoxic, non-marine system, there is no compelling reason to posit selection for the efficient use of macronutrients such as N and P; measured N concentrations near the periods when the metagenomic sequence data were collected were much higher than those of a typical N-limited marine system (Kenneth Hurst Williams, personal communication). However, the Rifle aquifer has been a site of intense bioremediation experimentation for some time, and it is possible that, before this activity, organisms had evolved with N or other nutrient limitation that drove the observed trend toward streamlined genomes.
Sabath et al. (2013) used comparative genomics to study streamlining in thermophiles and reported clear evidence that, in bacteria and archaea, genome size decreases with increasing optimum growth temperature. Their analysis indicates that neither genetic drift nor selection for short division times explains the strong overall trend to small genomes in extreme thermophiles. As with the aquifer microorganisms studied by Wrighton et al. (2012) and Kantor et al. (2013), there is no evidence to support the conclusion that reduction of the material costs of replication (that is, the amount of N and P required for a cell division) explains the observed trend. Sabath and co-workers did not explore the metabolic implications of streamlining, and so their work does not rule out that increasing nutritional requirements, implying increased connectivity in thermophile communities, might explain the trend to small genome size. They could not prove, but favored, the hypothesis that small cell size, and associated low maintenance energy costs, might be the driving force behind genome minimization in thermophiles.
Pelagibacter is the most thoroughly explored example of a streamlined chemoheterotrophic organism, and it illustrates how simple and efficient metabolic strategies can evolve to exploit nutrient resources that are part of the ambient background in biological systems. Pelagibacterales (SAR11) are the most abundant microbial group in the oceans worldwide, and are also common in freshwater (Giovannoni et al., 1990; Morris et al., 2002; Rusch et al., 2007; Carlson et al., 2009; Eiler et al., 2009; Schattenhofer et al., 2009; Treusch et al., 2009). They are obligate aerobes with full respiratory electron transport systems (Giovannoni et al., 2005; Grote et al., 2012) and the light-dependent proton pump proteorhodopsin, which has been shown to substitute as a power supply when organic carbon nutrients are unavailable from the environment to support respiration (Steindler et al., 2011). They have one rRNA operon, never grow faster than one division per day in culture, and are inhibited by many different organic compounds at levels far below those tolerated by copiotrophs (Rappé et al., 2002; Giovannoni et al., 2005).
Pelagibacter metabolism is spare, lacking most common auxiliary functions of microbial cells, and as discussed below, even lacking some functions that are nearly universally distributed among related Alphaproteobacteria (Giovannoni et al., 2005; Tripp et al., 2008, 2009; Carini et al., 2012; Grote et al., 2012; Smith et al., 2013). But these organisms are efficient oxidizers of a range of common, low-molecular-weight metabolites, including amino acids, organic acids and osmolytes. One of the surprises of Pelagibacter metabolism was the discovery that they devote part of their genome to one-carbon (C1) metabolism. C1 metabolism enables Pelagibacterales to oxidize a broad range of external metabolites, including C1 compounds, such as methanol and formaldehyde, and methylated compounds, including osmolytes (glycine betaine, taurine, 3-dimethyl-sulfoniopropionate and trimethylamine oxide; Sun et al., 2011). Osmolytes, also known as compatible solutes, are fundamental compounds in the sense that they are produced in large amounts by all cells in marine systems. The focus of Pelagibacterales on classes of organic compounds that are omnipresent, low-molecular-weight products of biological processes probably explains why they are a large proportion of all cells in the ocean throughout the water column. Microbial ecologists refer to the class of compounds used by Pelagibacterales cells as ‘labile dissolved organic matter’, which implies these compounds are turned over rapidly and rarely reach high concentrations. Thus, Pelagibacterales appear to have a simple life-cycle strategy that requires little regulation and depends on efficient competition for common organic compounds that flux continuously in active planktonic systems.
In contrast, relatively large microbial genomes enable organisms to search for resources (chemotaxis), change strategies to exploit patchy nutrients and target metabolites such as complex polysaccharides, which require complex enzyme repertoires. In long-term studies of population dynamics in the ocean, easily cultured copiotrophic organisms, such as members of the genera Alteromonas and Vibrio, are frequently rare but bloom stochastically (Vergin et al., 2013).
Unusual nutrient requirements related to genome streamlining may explain why many abundant microorganisms are challenging to culture
It is ironic that some of the best examples of streamlining are found in cultivated microorganisms. It is important to recognize that these organisms resisted cultivation by traditional techniques and were cultivated with specialized methods only after their molecular data showed they were important in nature. Success isolating chemoheterotrophic strains depended on the use of natural media made from autoclaved or filtered water samples, where naturally occurring compounds supplied organic matter for growth. This approach produces low population densities (about 106 cells per ml) but avoids growth inhibition and provides the opportunity to use insight gained from genomics to optimize cultivation conditions. Genome reduction has been directly linked to unusual growth requirements in Pelagibacter, explaining why these cells are challenging to culture. In addition to the aforementioned glycine auxotrophy (Tripp et al., 2009), they require reduced sulfur compounds (for example, dimethylsulfoniopropionate or methionine) because they lack the common assimilatory sulfate reduction pathway (Tripp et al., 2008). Recently, it was shown that Pelagibacter spp. require pyruvate, or pyruvate precursors, in a 4:1 ratio to glycine (Carini et al., 2012). This unusual example of stoichiometric nutrient co-dependence remains incompletely understood, but appears to be a consequence of the highly irregular arrangement of pathways in central metabolism. As streamlined cells appear to be common in nature, we hypothesize that unexpected growth requirements resulting from genome streamlining can explain a significant part of the ‘unculturable’ phenomenon.
In this issue of ISME J, we report a new and unusual example of a nutritional requirement caused by genome reduction in Pelagibacter. These cells are missing many thiamin biosynthesis, salvage and transport genes, and hence require the thiamin (vitamin B1) precursor 4-amino-5-hydroxymethyl-2-methylpyrimidine (HMP) (Carini et al., 2014).
Streamlining, co-evolutionary dynamics and ecological theory
Reports of high connectedness in microbial communities have garnered attention because connectedness can help predict the responses of biological communities to stresses (Faust and Raes, 2012). Connectedness is manifested as correlations between fluctuating populations, and can be due to interactions such as production of a compound by one organism and its use by another. Unusual nutrient requirements related to genome reduction can potentially explain some connectedness, and further, lead to predictions of co-evolutionary dynamics. As competition for resources increases, there is a concomitant rise in the relative cost of prototrophy (the ability to make a compound needed for growth). Thus, gene loss is favored when the advantage of needing less phosphorous, nitrogen- or carbon-derived energy to replicate outweighs the cost of sometimes being unable to replicate because an essential nutrient is unavailable.
The BQH invoked genome reduction and co-evolutionary models to explain nutritional requirements (Morris et al., 2012). This hypothesis argues that many vital metabolic pathways produce ‘leaky’ products—metabolites, or detoxification functions—which can escape from the cell and thus become public goods within a microbial community. In the case of leaked metabolites, organisms capable of transporting these metabolites across their cellular membrane no longer have to synthesize the metabolites de novo. Concomitant loss of genes involved in metabolite synthesis under streamlining provides a selective advantage under limited resources at the cost of auxotrophy for community resources. Thus, the BQH provides a compelling framework for a mechanism of streamlining and increasing community connectedness under nutrient limitation.
Beguiling though it may be, many examples of streamlining do not fit into the BQH. For example, rather than prototrophic loss, much of genome reduction in Pelagibacter, the most well-understood example, eliminates metabolic pathways and regulatory complexity that enable cells to adaptively respond to a wider range of conditions. For example, the first alphaproteobacterium PII-independent system for responding to nitrogen stress was recently described in Pelagibacter (Smith et al., 2013). This regulatory system, which involves transcriptional regulators as well as a number of novel, conserved riboswitch-like structures, is encoded by a highly reduced gene set, relative to the system found in other Alphaproteobacteria. Second, genome streamlining under the conditions of the BQH increases connectedness through increased auxotrophy for specific substrates. However, many of the unusual nutrient requirements that make Pelagibacter difficult to culture can be met by a variety of compounds. For example, methionine, cysteine, dimethylsulfoniopropionate or methanethiol can supply these cells with sulfur (Tripp et al., 2008 and unpublished results). Not surprisingly, in Pelagibacter, evolution has favored paths of genome reduction that minimize compromises to fitness that are associated with co-evolutionary dynamics. If streamlined organisms are as prevalent in the environment as now appears likely, and if the Pelagibacter model can be generalized, then we can expect to commonly find simplified versions of bacteria that are adapted to grow slowly but efficiently in a complex chemical milieu, rather than as a result of co-evolutionary connectedness between specific taxa.
Conclusions and Future prospects
Streamlining is a relatively new and important addition to microbial ecology theory. Ramifications of this theory are being explored by microbial ecologists who seek to explain the small genomes that are being discovered in a broad panorama of environments. Complex patterns of prototrophy associated with streamlining imply that some microbial communities have evolved high levels of nutritional connectivity that influence their stability. This same phenomenon also has the power to explain the prevalence of the uncultured microbial majority; it produced the testable hypothesis that uncultured microbial diversity is disproportionately populated with taxa that have streamlined genomes and therefore are difficult to culture. One highly studied example of streamlining, Pelagibacter, is providing insight into non-canonical rearrangements of metabolism that result in unexpected forms of prototrophy, including conditional prototrophy (nutritional requirements that are context dependent), and requirements for specific ratios of nutrients. Discoveries of alternate arrangements of genes into pathways, as with discoveries of new gene functions, can have a broad influence when they are applied retrospectively to interpret metabolic functions from genomic and metagenomic data.
Notwithstanding our enthusiasm for streamlining theory, we assure our readers that we do not think every example of odd cultivation requirements, connectedness in the environment or strange metabolism has an origin in streamlining selection. Neither are concepts like copiotroph and oligotroph or r- and k-strategist sufficient in themselves to categorize metabolic diversity. We propose that what is needed is: (1) research that more tightly links fundamental properties of cells, such as biomass, transport kinetics and metabolic efficiency to genome characteristics and (2) a better understanding of the adaptive strategies that link environmentally important species to the successful exploitation of particular resources. Systems biology, tailored to explain environmental theory, is an avenue that is relatively underexplored but appears to hold promise for addressing these issues. This was an approach that once applied only to model organisms, but today has conceptual appeal across a broad range of scales.
References
Angly FE, Willner D, Prieto-Davo A, Edwards RA, Schmieder R, Vega-Thurber R et al. (2009). The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput Biol 5: e1000593.
Babu MM . (2003). Did the loss of sigma factors initiate pseudogene accumulation in M-leprae? Trends Microbiol 11: 59–61.
Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD et al. (2010). Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci USA 107: 8806–8811.
Button DK . (1991). Biochemical basis for the whole-cell uptake kinetics: specific affinity, oligotrophic capacity, and the meaning of the michaelis constant. Appl Environ Microbiol 57: 2033–2038.
Button DK . (1998). Nutrient uptake by microorganisms according to kinetic parameters from theory as related to cytoarchitecture. Microbiol Mol Biol Rev 62: 636–645.
Button DK, Robertson BR . (2001). Determination of DNA content of aquatic bacteria by flow cytometry. Appl Environ Microbiol 67: 1636–1645.
Carini P, Campbell EO, Morré J, Sañudo-Wilhelmy SA, Thrash JC, Bennett SE et al. (2014). Discovery of a SAR11 growth requirement for thiamin's pyrimidine precursor and its distribution in the Sargasso Sea. ISME J doi:10.1038/ismej.2014.61.
Carini P, Steindler L, Beszteri S, Giovannoni SJ . (2012). Nutrient requirements for growth of the extreme oligotroph ‘Candidatus Pelagibacter ubique' HTCC1062 on a defined medium. ISME J 7: 592–602.
Carlson CA, Morris R, Parsons R, Treusch AH, Giovannoni SJ, Vergin K . (2009). Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J 3: 283–295.
Chisholm SW, Frankel SL, Goericke R, Olson RJ, Palenik B, Waterbury JB et al. (1992). Prochlorococcus marinus nov. gen. nov. sp.: an oxyphototrophic marine prokaryote containing divinyl chlorophyll a and b. Archive Microbiol 157: 297–300.
Dambach MD, Winkler WC . (2009). Expanding roles for metabolite-sensing regulatory RNAs. Curr Opin Microbiol 12: 161–169.
Dethlefsen L, Schmidt TM . (2007). Performance of the translational apparatus varies with the ecological strategies of bacteria. J Bacteriol 189: 3237–3245.
Dixon JL, Beale R, Nightingale PD . (2011). Rapid biological oxidation of methanol in the tropical Atlantic: significance as a microbial carbon source. Biogeosciences 8: 2707–2716.
Doolittle WF, Sapienza C . (1980). Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601–603.
Dufresne A, Garczarek L, Partensky F . (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol 6: R14.
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Alexander Richter R, Valas R et al. (2011). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Eiler A, Hayakawa DH, Church MJ, Karl DM, Rappe MS . (2009). Dynamics of the SAR11 bacterioplankton lineage in relation to environmental conditions in the oligotrophic North Pacific subtropical gyre. Environ Microbiol 11: 2291–2300.
Eiler A, Zaremba-Niedzwiedzka K, Garcia MM, McMahon KD, Stepanauskas R, Andersson SG et al. (2013). Productivity and salinity structuring of the microplankton revealed by comparative freshwater metagenomics. Environ Microbiol 2013; e-pub ahead of print 10 October 2013; doi:10.1111/1462-2920.12301.
Falkowski PG, Barber RT, Smetacek VV . (1998). Biogeochemical controls and feedbacks on ocean primary production. Science 281: 200–207.
Faust K, Raes J . (2012). Microbial interactions: from networks to models. Nat Rev Microbiol 10: 538–550.
Frank JA, Sorensen SJ . (2011). Quantitative metagenomic analyses based on average genome size normalization. Appl Environ Microbiol 77: 2513–2521.
Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP . (2009). The bacterial species challenge: making sense of genetic and ecological diversity. Science 323: 741–746.
Fry JC . (1990). Oligotrophs. In: Edwards, (ed) Microbiology of Extreme Environments. McGraw-Hill Publishing Company: New York, pp 93–116.
Ghai R, Mizuno CM, Picazo A, Camacho A, Rodriguez-Valera F . (2013). Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria. Sci Rep UK 3: 2471.
Giovannoni SJ, Britschgi TB, Moyer CL, Field KG . (1990). Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63.
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309: 1242–1245.
Giovannoni SJ, Hayakawa DH, Tripp HJ, Stingl U, Givan SA, Cho JC et al. (2008). The small genome of an abundant coastal ocean methylotroph. Environ Microbiol 10: 1717–1782.
Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ et al. (2012). Streamlining and core genome conservation among highly divergent members of the SAR11 clade. MBio 3: e00252–12.
Grzymski JJ, Dussaq AM . (2012). The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J 6: 71–80.
Halsey KH, Carter AE, Giovannoni SJ . (2012). Synergistic metabolism of a broad range of C1 compounds in the marine methylotrophic bacterium HTCC2181. Environ Microbiol 14: 630–640.
Hartigan JA, Hartigan PM . (1985). The dip test of unimodality. Ann Stat 13: 70–84.
Ishimoto KS, Lory S . (1989). Formation of pilin in Pseudomonas-Aeruginosa requires the alternative sigma-factor (Rpon) of Rna-polymerase. Proc Natl Acad Sci USA 86: 1954–1957.
Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4: e00708–e00713.
Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S et al. (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet 3: e231.
Kimura M . (1968). Evolutionary rate at the molecular level. Nature 217: 624–626.
Klappenbach JA, Dunbar JM, Schmidt TM . (2000). rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol 66: 1328–1333.
Koonin EV, Wolf YI . (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res 36: 6688–6719.
Kuo CH, Moran NA, Ochman H . (2009). The consequences of genetic drift for bacterial genome complexity. Genome Res 19: 1450–1454.
Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 106: 15527–15533.
Lynch M, Conery JS . (2003). The origins of genome complexity. Science 302: 1401–1404.
Lynch M . (2006). Streamlining and simplification of microbial genome architecture. Ann Rev Microbiol 60: 327–349.
Manganelli R, Dubnau E, Tyagi S, Kramer FR, Smith I . (1999). Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol 31: 715–724.
McCutcheon JP, Moran NA . (2012). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26.
Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ et al. (2009). Identification of candidate structured RNAs in the marine organism ‘Candidatus Pelagibacter ubique’. BMC Genomics 10: 268.
Mira A, Ochman H, Moran NA . (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet 17: 589–596.
Morris JJ, Johnson ZI, Szul MJ, Keller M, Zinser ER . (2011). Dependence of the cyanobacterium Prochlorococcus on hydrogen peroxide scavenging microbes for growth at the ocean's surface. PLoS One 6: e16805.
Morris JJ, Lenski RE, Zinser ER . (2012). The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 3: e00036–12.
Morris RM, Longnecker K, Giovannoni SJ . (2006). Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ Microbiol 8: 1361–1370.
Morris RM, Rappe MS, Connon SA, Vergin KL, Siebold WA, Carlson CA et al. (2002). SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420: 806–810.
Moya A, Gil R, Latorre A, Pereto J, Pilar Garcillan-Barcia M, de la Cruz F . (2009). Toward minimal bacterial cells: evolution vs. design. FEMS Microbiol Rev 33: 225–235.
Oh S, Caro-Quintero A, Tsementzi D, DeLeon-Rodriguez N, Luo C, Poretsky R et al. (2011). Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl Environ Microbiol 77: 6000–6011.
Orgel LE, Crick FH . (1980). Selfish DNA: the ultimate parasite. Nature 284: 604–607.
Partensky F, Garczarek L . (2010). Prochlorococcus: advantages and limits of minimalism. Annu Rev Mar Sci 2: 305–331.
Quaiser A, Zivanovic Y, Moreira D, Lopez-Garcia P . (2011). Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J 5: 285–304.
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P . (2007). Prediction of effective genome size in metagenomic samples. Genome Biol 8: R10.
Ranea JA, Grant A, Thornton JM, Orengo CA . (2005). Microeconomic principles explain an optimal genome size in bacteria. Trends Genet 21: 21–25.
Rappé MS, Connon SA, Vergin KL, Giovannoni SJ . (2002). Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418: 630–633.
Rocap G, Distel DL, Waterbury JB, Chisholm SW . (2002). Resolution of Prochlorococcus and Synechococcus ecotypes by using 16S-23S ribosomal DNA internal transcribed spacer sequences. Appl Environ Microbiol 68: 1180–1191.
Rocap G, Larimer FW, Lamerdin J, Malfatti S, Chain P, Ahlgren NA et al. (2003). Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424: 1042–1047.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al. (2007). The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol 5: 398–431.
Sabath N, Ferrada E, Barve A, Wagner A . (2013). Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol Evol 5: 966–977.
Schattenhofer M, Fuchs BM, Amann R, Zubkov MV, Tarran GA, Pernthaler J . (2009). Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ Microbiol 11: 2078–2093.
Smith DP, Kitner JB, Norbeck AD, Clauss TR, Lipton MS, Schwalbach MS et al. (2010). Transcriptional and translational regulatory responses to iron limitation in the globally distributed marine bacterium Candidatus pelagibacter ubique. PLoS One 5: e10487.
Smith DP, Thrash JC, Nicora CD, Lipton MS, Burnum-Johnson KE, Carini P et al. (2013). Proteomic and transcriptomic analysis of Candidatus Pelagibacter ubique describes the first PII-independent response to nitrogen limitation in a free-living alphaproteobacterium. MBio 4: e00133–12.
Sowell SM, Abraham PE, Shah M, Verberkmoes NC, Smith DP, Barofsky DF et al. (2011). Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 5: 856–865.
Sowell SM, Wilhelm LJ, Norbeck AD, Lipton MS, Nicora CD, Barofsky DF et al. (2008). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J 3: 93–105.
Steglich C, Futschik ME, Lindell D, Voss B, Chisholm SW, Hess WR . (2008). The challenge of regulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet 4: e1000173.
Steindler L, Schwalbach MS, Smith DP, Chan F, Giovannoni SJ . (2011). Energy starved Candidatus Pelagibacter ubique substitutes light-mediated ATP production for endogenous carbon respiration. PLoS One 6: e19725.
Sun J, Steindler L, Thrash JC, Halsey KH, Smith DP, Carter AE et al. (2011). One carbon metabolism in SAR11 pelagic marine bacteria. PLoS One 6: e23973.
Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, Gonzalez JM et al. (2013). Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci USA 110: 11463–11468.
Thingstad TF, Øvrea L, Egge JK, Løvdal T, Heldal M . (2005). Use of non-limiting substrates to increase size;a generic strategy to simultaneously optimize uptake and minimize predation in pelagic osmotrophs? Ecol Lett 8: 675–682.
Touchon M, Rocha EP . (2007). Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol 24: 969–981.
Treusch AH, Vergin KL, Finlay LA, Donatz MG, Burton RM, Carlson CA et al. (2009). Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J 3: 1148–1163.
Tripp HJ, Kitner JB, Schwalbach MS, Dacey JW, Wilhelm LJ, Giovannoni SJ . (2008). SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452: 741–744.
Tripp HJ, Schwalbach MS, Meyer MM, Kitner JB, Breaker RR, Giovannoni SJ . (2009). Unique glycine-activated riboswitch linked to glycine-serine auxotrophy in SAR11. Environ Microbiol 11: 230–238.
van Ham RC, Kamerbeek J, Palacios C, Rausell C, Abascal F, Bastolla U et al. (2003). Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci USA 100: 581–586.
Vergin KL, Tripp HJ, Wilhelm LJ, Denver DR, Rappe MS, Giovannoni SJ . (2007). High Intraspecific Recombination Rate in a Native Population of Candidatus Pelagibacter ubique (SAR11). Environ Microb 9: 2430–2440.
Vergin KL, Done B, Carlson CA, Giovannoni SJ . (2013). Spatiotemporal distributions of rare bacterioplankton populations indicate a variety of adaptive strategies in the oligotrophic ocean. Aquat Microb Ecol 71: 1–13.
Vieira-Silva S, Rocha EP . (2010). The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet 6: e1000808.
Vieira-Silva S, Touchon M, Rocha EP . (2010). No evidence for elemental-based streamlining of prokaryotic genomes. Trends Ecol Evol 25: 319–320 author reply 320-311.
Waters LS, Storz G . (2009). Regulatory RNAs in bacteria. Cell 136: 615–628.
Wilhelm LJ, Tripp HJ, Givan SA, Smith DP, Giovannoni SJ . (2007). Natural variation in SAR11 marine bacterioplankton genomes inferred from metagenomic data. Biol Direct 2: 27.
Winkler WC, Breaker RR . (2005). Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol 59: 487–517.
Wösten MM . (1998). Eubacterial sigma-factors. FEMS Microbiol Rev 22: 127–150.
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665.
Xia LC, Cram JA, Chen T, Fuhrman JA, Sun F . (2011). Accurate genome relative abundance estimation based on shotgun metagenomic reads. PLoS One 6: e27992.
Yoshimura T, Imamura S, Tanaka K, Shirai M, Asayama M . (2007). Cooperation of group 2 sigma factors, SigD and SigE for light-induced transcription in the cyanobacterium Synechocystis sp PCC 6803. FEBS Lett 581: 1495–1500.
Acknowledgements
We are grateful to an anonymous reviewer, whose many critical but helpful comments made this review much better. This work was supported by a grant from the Marine Microbiology Initiative of the Gordon and Betty Moore Foundation (to SJG), NSF Microbial Observatory grant no. OCE-0802004 (to SJG), a Community Sequencing Project grant (csp2009.797268) and NSF award DBI-1003269 (to JCT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Giovannoni, S., Cameron Thrash, J. & Temperton, B. Implications of streamlining theory for microbial ecology. ISME J 8, 1553–1565 (2014). https://doi.org/10.1038/ismej.2014.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.60
Keywords
This article is cited by
-
Metagenomic characterization of a novel non-ammonia-oxidizing Thaumarchaeota from hadal sediment
Microbiome (2024)
-
Pseudogenes act as a neutral reference for detecting selection in prokaryotic pangenomes
Nature Ecology & Evolution (2024)
-
Genomic analyses reveal a low-temperature adapted clade in Halorubrum, a widespread haloarchaeon across global hypersaline environments
BMC Genomics (2023)
-
The role of the ecological scaffold in the origin and maintenance of whole-group trait altruism in microbial populations
BMC Ecology and Evolution (2023)
-
Metagenome-assembled genomes reveal greatly expanded taxonomic and functional diversification of the abundant marine Roseobacter RCA cluster
Microbiome (2023)
