
Abstract
Permafrost soils are large reservoirs of potentially labile carbon (C). Understanding the dynamics of C release from these soils requires us to account for the impact of wildfires, which are increasing in frequency as the climate changes. Boreal wildfires contribute to global emission of greenhouse gases (GHG—CO2, CH4 and N2O) and indirectly result in the thawing of near-surface permafrost. In this study, we aimed to define the impact of fire on soil microbial communities and metabolic potential for GHG fluxes in samples collected up to 1 m depth from an upland black spruce forest near Nome Creek, Alaska. We measured geochemistry, GHG fluxes, potential soil enzyme activities and microbial community structure via 16SrRNA gene and metagenome sequencing. We found that soil moisture, C content and the potential for respiration were reduced by fire, as were microbial community diversity and metabolic potential. There were shifts in dominance of several microbial community members, including a higher abundance of candidate phylum AD3 after fire. The metagenome data showed that fire had a pervasive impact on genes involved in carbohydrate metabolism, methanogenesis and the nitrogen cycle. Although fire resulted in an immediate release of CO2 from surface soils, our results suggest that the potential for emission of GHG was ultimately reduced at all soil depths over the longer term. Because of the size of the permafrost C reservoir, these results are crucial for understanding whether fire produces a positive or negative feedback loop contributing to the global C cycle.
Similar content being viewed by others

Introduction
Fire is a major factor controlling the long-term dynamics of soil C and permafrost stability in boreal ecosystems (Harden et al., 2000; Schuur et al., 2008; Jorgenson et al., 2013). Although boreal ecosystems are fire-adapted, increased fire frequency (Randerson et al., 2006) due to climate change has altered the age structure of the vegetation (old to young) and its recovery (Czimczik et al., 2006). Burning biomass contributes to global emissions of CO2; the intense heat and the modified surface conditions results in a deepening of the active layer and thawing near-surface permafrost (Johnstone et al., 2010; Nossov et al., 2013). During a fire, the insulating organic surface layer is often destroyed and eventually mosses regrow. A major concern is that with a warmer and drier climate, increased fire frequency and intensity may trigger a positive feedback loop between the loss of soil C and subsequent warming and thawing of permafrost soils (McGuire et al., 2007; O'Donnell et al., 2011). Over longer time scales, fire-based disturbance can lead to major shifts in a variety of environmental parameters that are likely to have large direct and indirect effects on the soil microbial community; however, these shifts include C sequestration in refractory biochar, which is not available to microbial metabolism and which may actually reduce C emissions in the longer term (Lehmann et al., 2009). Because the size of the C reservoir in permafrost is estimated to be 1672 Pg (Tarnocai et al., 2009)—roughly equal to the total amount in vegetation and the atmosphere—it is critical that we understand the balance between long- and short-term effects to predict whether fire is a positive (via increased green house gas (GHG) emissions) or a negative (via accumulation of pyrogenic C) feedback in global C cycling.
It has been predicted that global warming could thaw 25% of the Arctic permafrost area by 2100, exposing large amounts of currently fixed organic C to microbial decomposition (Coolen et al., 2011). At fire-impacted sites where vegetation regeneration is slow, rising temperatures may increase the metabolic rates of decomposer microbes and long-term permafrost degradation may continue (Jorgenson et al., 2010). The rate of organic matter decomposition and CO2 release from fire-affected boreal soils and permafrost has only recently been investigated (Waldrop and Harden, 2008; Turetsky et al., 2011). Although postfire soils can potentially release more CO2 to the atmosphere under elevated temperatures (Bergner et al., 2004), positive feedbacks between CO2 release and soil warming may not be uniform or sustained (O’Donnell et al., 2009; Allison et al., 2010). This is particularly problematic when warming causes more consistent C losses in regions underlain by permafrost (Schuur et al., 2008).
Fire-mediated changes in the soil microbial community structure are currently unknown, but are likely to have consequences for nutrient cycling and related processes. Microbial decomposition rates have previously been reported to decline after fire. However, even low decomposition rates from microbial respiration can still cause significant C losses after fire (Bond-Lamberty et al., 2004). Defining the key microbial players involved in residual C decomposition in burned boreal ecosystems is important to understanding their contribution to the global CO2 budget. Here our aim was to study the impact of fire on microbial community structure and function in upland boreal forests with a specific focus on GHG emissions and microbial C cycling processes. We studied the fire impact on both surface and permafrost zones to detect responses to fire at different depths in the soil profile.
The sampling location near Nome Creek, Alaska was affected by the Boundary Fire that burned continuously from June to August 2004. The region is within the discontinuous permafrost zone, with approximately 75–80% of the ground underlain by permafrost starting from an approximate depth of 0.50 m (Osterkamp and Romanovsky, 1999). After the fire, both the surface organic and deeper mineral soil layers were 2–5 °C warmer, and winter freezing of the deeper mineral soil layers, which were previously permafrost, no longer occurred (Nossov et al., 2013). Here we collected samples from both burned and adjacent unburned locations and used a combination of extensive geochemical characterization, metagenome sequencing and enzymatic assays to determine which soil properties and microbial processes were affected by the fire. These data were used to inform the impact of fire on permafrost-affected soils in boreal regions on potential GHG production rates.
Materials and methods
Site selection and sample collection
The Nome Creek area is located ∼100 km north east of Fairbanks, AK and in the southeastern portion of the White Mountains National Recreation Area. This region is in the subpolar continental climatic zone, which is characterized by long, cold winters and short, hot summers with temperature extremes ranging from near –37 °C in winter to +20 °C in summer. The annual precipitation averages 55–75 mm and the average surface soil moisture varies between 10–50% (Kelsey et al., 2012). The Nome Creek area is predominantly covered by black spruce (Picea mariana) forests, which has an understory dominated by ericaceous shrubs, feathermosses and sphagnum (Nossov et al., 2013). Mean annual surface (−5 cm) temperature at the unburned sites was 0.5 °C vs 2.5 °C in the burned sites, and mean annual deep (−100 cm) temperature in the unburned and burned sites were −0.8 °C and 1.3 °C, respectively (Nossov et al., 2013). In September 2011, 7 years after the Boundary Fire, we collected replicate samples from three south-facing unburned and two adjacent burned locations (65.3452° latitude, −146.919° longitude). The organic horizon depth in the unburned soils was 42±4 cm, whereas at the burned site it was 11±9 cm. Over a broader area, the Boundary Fire consumed approximately 25 cm of surface organic matter from the forest floor (see Supplementary Table S5 in Turetsky et al., 2010).
Each sampling location was excavated up to a meter in depth and samples were collected every 10 cm. Soil surface was cleaned with sterile spatulas to remove debris and each pit was sampled from bottom to top to avoid cross-contamination from upper layers. Samples were stored and shipped on dry ice after collection, and stored at −80 °C until analysis. In the control locations, different sampling depths were grouped as surface (upper 40 cm of active layer), middle (40–60 cm in depth corresponding to lower active layer and upper part of permafrost; in this depth, soils may be thawed to 40–50 cm in cold summers and to 40–60 cm in warm years) and permafrost (permanently frozen with excess segregated ice) layers. In the wildfire-affected soils, the permafrost layer thawed throughout the depth of the measured profile. Therefore, 60–100 cm was referred to as deep soil. In September 2011, surface soil temperatures were similar (4–5 °C) in both burned and unburned locations, but in contrast to surface measurements, soil temperatures at 100 cm depth differed for each location (4 °C and −1 °C to 0 °C for burned and unburned areas, respectively) (Nossov et al., 2013).
Chemical characterization
All soil measurements were performed on homogenized root-free soil. Soil C and N were measured on a LECO C&N analyzer (LECO Corp., St Joseph, MI, USA). Soil pH and electrical conductivity (EC) were measured on soil slurries using standard pH and EC meters. Soil moisture was measured as the change in weight after 48 h drying at 105 °C. Dissolved organic carbon (DOC) was measured on 5 g soil extracted in 25 ml nanopure water for 24 h at 3 °C. Extracts were prefiltered under vacuum over glass fiber and then secondarily filtered through 0.45 μM nylon before analysis using a Shimadzu TOC-V CPH organic carbon analyzer (Shimadzu Corp., Kyoto, Japan). All samples were run in duplicate, averaged and blank corrected before expression per unit dry soil. Standards were run using 0–40 p.p.m. KH phthalate.
The chemical composition of organic matter in the soil samples and changes that occurred after the fire event were studied by analyzing the spectra obtained from solid state 13C-nuclear magnetic resonance (NMR). The 13C-NMR spectra for samples in 10 cm intervals from 0 to 30 cm in depth from one control and duplicate burned locations were obtained using a Varian Infinity CMX 300 MHz spectrometer (Varian NMR, Fort Collins, CO, USA). Samples were processed and measured according to previously published methods (Berhe et al., 2012). Briefly, dried and homogenized samples were packed into 5-mm zirconia rotors and fitted with boron-nitride spacers and KEL-F (low carbon) caps. We used a ramped (13C pulse) cross-polarization magic-angle spinning pulse sequence with a contact time of 1 ms, spinning rate of 10 kHz and a decoupling field of 63–55 kHz. The 13C chemical shifts were referenced to tetramethylsilane (0 p.p.m.) using an external reference, hexamethylbenzene (16.81 p.p.m.). Up to 30 000 scans were obtained for each sample and the data were processed using Fourier transformation, and a line broadening of 100 Hz was applied to all samples. The chemical fingerprint of C composition in soil was inferred by dividing the 13C-NMR spectra into seven chemical shift regions that correspond to seven functional groups, such as alkyl-C (0–45 p.p.m.), N-alkyl, methoxyl (45–60 p.p.m.), O-alkyl-C (60–95 p.p.m.), di-O-alkyl (95–110 p.p.m.); aromatic-C (110–160 p.p.m.); phenolic (145–165 p.p.m.) and carboxyl-C (165–215 p.p.m.).
Soil enzymes
Enzyme activity potentials involved in the extracellular decomposition of organic molecules, namely (hemi)cellulose, lignin, chitin, proteins and organic phosphate, were assayed on subsamples from all soil depths using artificial fluorogenic model substrates. The enzymes, their functions and substrates are listed in Supplementary Table S1. Briefly, potential activities of α-glucosidase, β-glucosidase (BG), β-cellobiohydrolase (CBH), β-xylosidase (XYL), N-acetyl glucosaminidase, phenol oxidase, peroxidase, acid phosphatase (P) and two peptidases (ALA, LEU) were screened (all substrates were purchased from Sigma-Aldrich, St Louis, MO, USA). Substrate concentrations were 200 mM. Phenol oxidase and peroxidase were assayed using L-dihydroxyphenylalanine as a substrate. Peroxidase also received 0.3% H2O2 as a cosubstrate. Substrates were made in 50 mM acetate–25 mM EDTA buffer at pH 5.5. Soil slurries were made by adding 1 g sample to 40 ml buffer at 4 °C and rapidly mixing (10 min) on a magnetic stir plate while pipetting. Assays were conducted by adding 500 μl substrate solution to 500 μl of soil slurry. Substrate controls and buffer controls were analyzed at the same time. 4-Methylumbelliferone and 7-amino-4-methylcoumarin were used to generate the standard curves. Standards were mixed and incubated with soils to account for possible sorption of these substrates to matrix. The 2 ml Costar 96-well plates (Corning Costar Corp., Cambridge, MA, USA) were covered and incubated either at 4 °C or at 15 °C for 24 h with occasional gentle mixing. After incubation, reaction plates were centrifuged 1 min at 1500 r.p.m. Two hundred microliters of the supernatant was transferred to reading plates where fluorescence or absorbance (for phenol oxidase and peroxidase) were measured using the Molecular Devices Gemini XS Fluorescent Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). Rates represent the mean of 16 technical replicates and presented as the amount of product evolved per unit time per gram soil (dry weight) or soil C.
GHG flux measurements
GHG flux measurements were carried out with soil samples from 10 to 20, 50 to 60 and 90 to 100 cm depths that were incubated under both aerobic and anaerobic conditions (n=2 per factorial combination of atmosphere × depth). Samples were stored frozen before incubation, but then thawed and homogenized under anaerobic conditions. One to five grams of homogenized soil samples were incubated under both aerobic and anaerobic conditions at 20 °C for 5 weeks. In all incubations, soil moisture was kept constant during the incubation period by adding water as necessary. Incubations under anaerobic conditions were conducted in 100 ml vials with blue stoppers and maintained by flushing the headspace with Ar after each weekly measurement. Incubations under aerobic conditions were sealed with 0.02 mm plastic wrap (Gordon et al., 1987) to prevent moisture loss, but permit diffusion between ambient atmosphere and incubation units between the headspace measurements. Once a week CO2, CH4 and N2O fluxes from soils were measured in the headspace using an SRI gas chromatograph (SRI 8610) with flame ionization detector, electron capture detector and methanizer (SRIGC, Torrance, CA, USA). First time-point measurement was made 72 h after the thawing of samples.
16S rRNA gene sequencing and analysis
Total DNA was extracted in duplicate from each sample by using 2 g as input into the PowerSoil DNA Isolation Kit (Mo Bio, Carlsbad, CA, USA) with minor modifications as described below. Before bead-beating, the samples were incubated in bead-solution at 65 °C for 5 min. Cells in the samples were disrupted by bead beating with the FastPrep Instrument (Qbiogene, Carlsbad, CA, USA) at a setting of 5.5 for 45 s and the DNA was further purified according to the kit protocol. DNA amounts were quantified using the Qubit dsDNA HS assay (Invitrogen, Carlsbad, CA, USA). 16S rRNA genes were amplified in polymerase chain reactions (PCRs) using primers (F515/R806) that target the V4 region of the 16S rRNA gene. The reverse PCR primer was barcoded with a 12-base Golay code (Caporaso et al., 2010). The PCR reactions contained 2.5 μl Takara Ex Taq PCR buffer, 2 μl Takara dNTP mix, 0.7 μl Roche bovine serum albumin (20 mg ml−1), 0.5 μl each of the forward and reverse primers (10 μM final concentration), 0.125 μl Takara Ex Taq Hot Start DNA Polymerase (TaKaRa, Shiga, Japan), 1.0 μl genomic DNA (5 ng per reaction) and nuclease-free water in total volume of 25 μl. Reactions were held at 98 °C for 3 min to denature the DNA, followed by amplification for 25 cycles at 98 °C for 30 s, 58 °C for 30 s and 72 °C for 45 s; a final extension of 10 min at 72 °C was added to ensure complete amplification. Each sample was amplified in triplicate, combined and purified using the Agencourt AMPure XP PCR purification system (Beckman Coulter, Brea, CA, USA). The purified amplicons were quantified using the Qubit dsDNA HS assay and the size of the amplicons was determined using a Bioanalyzer with Agilent DNA 1000 chips (Agilent Technologies, Santa Clara, CA, USA). Amplicons were pooled (25 ng per sample) and sequenced on one lane of the Illumina HiSeq 2000 platform (Illumina Inc., San Diego, CA, USA), resulting in 100 bp reads. Quality filtering and demultiplexing were performed as described previously (Caporaso et al., 2010; Bokulich et al., 2012). Quality data were not obtained from two samples (one of each replicated sample from one control location at 70–80 cm and one burn location at 30–40 cm) and these were excluded from further analyses. Operational taxonomic units (OTUs) were assigned from the reads using an open-reference OTU picking protocol against the Greengenes database release 12_10 clustered at 97% identity with UCLUST and assigned to a taxonomic unit with the RDP classifier (Wang et al., 2007) using QIIME (Caporaso et al., 2010) version 1.5.0. Phylogenetic trees were created using FastTree2 (Price et al., 2009) under QIIME’s default parameters and these trees were used for the calculation of α- (Shannon's H', Fisher’s α and Faith’s PD) and β-diversity (weighted UniFrac distance) metrics. Communities were standardized to a total number of 141 000 sequences per sample. The weighted UniFrac distance matrices were used to visualize the community composition, with depth through ordination in a correspondence analysis.
Metagenome sequencing and annotation
Twelve metagenomic shotgun sequencing libraries were prepared representing duplicate locations: control and burned sites and different depths along the soil profiles (10–20, 50–60 and 90–100 cm). Total genomic DNA (500 ng) was extracted from these samples as described above and sheared using the Covaris S-Series instrument (Covaris, Woburn, MA, USA). Sequencing libraries were prepared using a Illumina TruSeq DNA Sample Preparation Kit version 2 (Low-Throughput protocol) (Illumina Inc.) according to the manufacturer’s instructions and sequenced using the Illumina HiSeq 2000 single-read technology (at the Yale Center for Genome Analysis, CT, USA). Three samples were multiplexed in one lane of Illumina HiSeq 2000, resulting in 9.1–13.7 Gb of high-quality (minimum quality cutoff of 3) sequence data of 92 bp in length (Supplementary Table S3). Sequences were deposited at NBCI (BioProject ID: PRJNA223407).
A de novo comparative metagenomic analysis algorithm, Compareads, was used to compute the pairwise similarity measures between metagenomics data sets (Maillet et al., 2012). Two reads were assumed to be similar if they shared at least two k-mers of 33 nucleotides. The Compareads approach is sensitive to the number of reads used in the analysis, where imbalance in the number of sequences per sample results in false similarity estimates. The minimum number of reads was set to be 5.8 × 107, and because of low read depths for one of the samples (Burn: 10–20 cm), it was excluded from further Compareads analysis. Prodigal (Hyatt et al., 2010) was used to predict coding regions from the single reads. The translated proteins from all detected coding regions of each metagenome were annotated by searching against a custom in-house database (Prestat et al., in review) of hidden Markov models generated from Pfam profiles (Release 26.0; Finn et al., 2010). The FOAM (Functional Ontology Assignments for Metagenomes) database is publically available at (http://portal.nersc.gov/project/m1317/FOAM/). Microbial functions that contribute to soil biogeochemical cycles were inventoried and subsequently 617 were selected corresponding to KEGG Orthology (KO) (Kanehisa et al., 2008) -based classifications. This database contained the KO’s for the functions coding for: ribonucleases, RNA polymerases, carbohydrate metabolism, carbon fixation, nitrogen metabolism (nitrogen fixation, nitrification, denitrification, assimilatory nitrate reduction, dissimilatory nitrate reduction), methane oxidation, methanogenesis, sulfur reduction, oxidoreductases and stress responses. A total of 20 473 hidden Markov models were built upon sequence profiles very specific to each KO. The relative abundance of sequences was represented as a ratio of total gene counts for each KO to recombinase A gene copies in each metagenome. In addition, single reads were assembled using the CLC Genomics Workbench (version 5.0.1; CLC Bio, Cambridge, MA, USA) de novo assembler by following parameters: mismatch cost 2, insertion cost 3, deletion cost 3, length fraction 0.5 and similarity 0.8. The minimum contig length was set to 200 bp.
Data analysis
Statistical comparisons for soil chemical analyses were performed in JMP (SAS Institute, Cary, NC, USA). Repeated-measures analysis of variance (ANOVA) was used on time-series C mineralization measurements, with site (control vs burned), depth and atmosphere (aerobic vs anaerobic) as independent factors and CO2 flux as the dependent factor. Two-way ANOVA (site × depth) was used to test for differences in soil pH, soil EC and soil C and N. All other statistical tests were produced by using R packages (ade4 (Chessel et al., 2012) and vegan (Oksanen et al., 2007)) packages in the R statistical environment (http://www.r-project.org). Results are defined to be significant at P<0.05. To test differences in enzyme activity in the control vs burned samples, one-way ANOVA followed by the Tukey’s honestly significant difference test was used. Ordination of the whole community detected by 16S rRNA gene amplicon sequencing was created from UniFrac matrix calculated by QIIME software and presented in a principal coordinates analysis plot. The contribution of sampling strategy (fire × depth) and soil geochemistry to the observed variation in β-diversity was tested via variation partitioning and subsequent ANOVA analysis. Furthermore, principal component analysis (PCA) was performed on Hellinger transformed relative OTU abundances (downweights highly abundant OTUs while avoiding overweighting of rare OTUs) (Legendre and Gallagher, 2001; Ramette, 2007). To determine which environmental parameter (i.e. depth, burn, soil geochemistry and total enzyme activity) could significantly explain the variation in bacterial community structure, a canonical redundancy analysis was used, and its significance was assessed by ANOSIM (analysis of similarities; 999 Monte Carlo permutation tests). In cases where the redundancy analysis achieved statistical significance, Pearson’s correlation tests were used to detect significant positive or negative correlations of each OTU abundance distribution with the corresponding environmental variable. Pairwise similarities calculated by Compareads between the metagenomes were visualized by heatmaps and hierarchical clustering. Normalized KO relative abundances were used to explore the correlations between different depths of fire-impacted and control samples. Between-class analysis was used as implemented in ade4 package, where the analysis finds the principal components based on the center of gravity of each group. Significance of this analysis was tested with a subsequent ANOVA. Differences in the relative abundances of C- and N-cycle genes were represented as a heatmap, where data matrix was centered by subtracting the column means and scaled by dividing the (centered) columns by their standard deviation.
Results
Physical and chemical characterization
The stratigraphic profile of the soil at the Nome Creek control locations had an organic layer depth of 42±4 cm and a permafrost layer starting at 46±1 cm in depth (Figure 1a). The pH increased with depth and ranged between 3.7 and 5.6 (Table 1). The active layer soils were acidic, whereas the permafrost layers had significantly higher pH values (P<0.001). Total C, N, DOC and moisture content declined with increasing depth, with the active layer characterized by high C and water contents (Table 1).

(a) Photos showing the intact and thawed permafrost soils of Nome Creek. Tick marks correspond to 10 cm intervals. (b) CO2 fluxes in the headspace of 8-week long laboratory incubation of samples from control and burned locations. Values are means±s.e. (c) Changes in the extracellular enzyme activities between control and burned locations.
The conditions were very different for the adjacent burned locations. The surface organic horizon was 11±9 cm thick, indicating that the fire burned the top ∼30 cm of the surface organic material. In addition, the permafrost was completely thawed over the measured depth of 1 m. The fire-impacted soils had a significantly higher pH (P<0.05) and reduced soil C, N, DOC and moisture contents compared with controls (Table 1). In the top 50 cm, 50–90% of soil C and 86–96% of the soil DOC were depleted after the fire. However, when controlling for the consumption of surface organic horizons, fire-impacted surface soils (0–30 cm) had similar C or N concentrations to mineral layers of control locations (30–50 cm). This comparison suggested that there was no residual effect of burning on soil C or N concentrations in mineral soils. However, DOC concentrations were significantly (P<0.05) lower in burned soils compared with control soils even after controlling for surface organic horizon loss.
13C-NMR analysis was used to provide detailed information about the organic C composition in duplicate samples (Supplementary Figure S1A) obtained at different active layer depths reported to be impacted by the fire: 0–30 cm, in 10 cm depth intervals. Aromacity (%), as a measure of the largely aromatic pyrogenic C accumulation after the fire, was higher in the surface 0–10 cm layer of the burned plots compared with all layers in the control plots and deeper layers of the burned plots (Supplementary Figure S1B), suggesting that the fire had the most impact on the surface organic matter composition. The alkyl/O–alkyl ratio of soil organic matter (SOM) suggests that SOM in the burned plots is more transformed by oxidative decomposition. The higher alkyl/O-alkly ratio suggest that easily decomposable sugars, simple amino acids and alkyls (such as lipids) were depleted due to decomposition (Berhe et al., 2012). The alkyl/O–alkyl ratio was also higher below the surface at the 10–30 cm depths. The ratio of oxidized C functional groups compared with the ring structures (the COOH/aromatic ratio) was highest at the mid-depth of 10–20 cm.
GHG fluxes
Using repeated-measures ANOVA on 8 weeks of CO2 flux data from aerobic incubations (Figure 1b) showed a significant site × depth interaction (F=58.416, P<0.001). The interaction is explained by significantly (P<0.001) higher aerobic respiration in the surface soil of the unburned site compared with all other soils. Thus, there were no differences in CO2 flux between burned and control soils below the surface soil and there were also no significant differences in CO2 fluxes with depth within the burned site. Flux data from anaerobic incubations showed a significant site × depth interaction (F=7.905, P<0.001) in CO2 emissions (Supplementary Figure S2A), where this interaction was significantly (P<0.001) explained by the marginal increase in CO2 fluxes from the control surface soils. Methane flux (Supplementary Figure S2B) was detected in middle and permafrost layers of the control locations and was highly variable. In burned locations, CH4 fluxes were significantly reduced (F=4.612, P=0.012). Low amounts of N2O emission (Supplementary Figure S2C) was detected in unburned surface soils; however, there was no significant difference in N2O levels between control and burned soils in any depth.
Potential soil enzyme activities
At both control and burned locations, the potential enzyme activities per gram soil decreased with depth (Supplementary Figure S3), but when the soil C content was taken into account, the enzyme activities per gram soil C were not significantly different (F=0.612, P=0.782) across the soil profiles in each location (Supplementary Table S2 and Figure 1c). Two temperatures (4 °C and 15 °C) were used to stimulate the enzyme activities; however, no significant difference in enzyme activities was observed within control and burned locations owing to different incubation temperatures. The highest enzyme activities were observed for cellulose- and chitin-degrading enzymes, whereas lignocellulose-degrading enzymes had low potential activities at all depths (results not shown). In control samples, neither carbohydrate-active (α-glucosidase, BG, CBH, XYL, N-acetyl glucosaminidase) nor aminopeptidase enzyme activities were significantly different between different soil depths. Only phosphatase activity was significantly (F=11.174, P<0.001) higher in surface soils in comparison with middle and permafrost layers. Fire caused a marginal increase in potential enzymatic activities at the surface (Figure 1c and Supplementary Table S2). In surface soils, enzymes contributing to the degradation of cellulose (CBH and BG), hemicellulose (XYL) and aminopeptidases had significantly higher activity among burned vs unburned areas (Supplementary Table S2). By contrast, in middle and deep soils there were no significant differences between control and burned samples. When the differences in soil depth were taken into account (by comparing enzyme activities in mineral layers of the surface soils), carbohydrate-active (BG, CBH, XYL, N-acetyl glucosaminidase), aminopeptidase and phosphatase activities were significantly higher in burned surface soils (Supplementary Table S2).
Microbial community composition and diversity
Wildfire had no significant impact on the α-diversity of the soils at all depths. After rarefaction to the same level of surveying effort (141 000 sequences per sample), we found 82 different family level assignments corresponding to a total of 202 OTUs across all samples. Owing to the high depth of sequencing, the rarefaction curves (Supplementary Figure S4) showed a high coverage approaching saturation in many of the samples. The number of OTUs remained similar with depth in both control and burned locations. Pairwise comparisons of α-diversity (species richness (Chao 1), species abundance (Fisher’s α), species diversity (Shannon’s H′) and phylogenetic diversity (Faith’s PD)) revealed significant changes in microbial diversity between different soil depths but not with fire (Supplementary Figures S5A and B). Species diversity (H′) was significantly higher in surface soils of both control (F=16.05, P<0.001) and burned locations (F=4.70, P<0.001) in comparison with permafrost and deep soils.
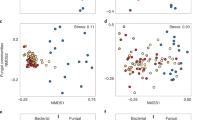
Although the communities primarily clustered by sampling depth (Figure 2a), fire also had a significant impact on the microbial β-diversity. We took into account the change in depth of the organic layer, due to consumption by fire, when interpreting the results. A principal coordinates analysis plot based on UniFrac distances could explain 58% of the observed variation (represented in the first two axes), where the first axis was correlated with depth (r2=0.43, P<0.001). The ordination observed in this axis was mainly driven by the differences between organic and mineral soil horizons (P=0.029). The second axis mainly correlated with moisture content (r2=0.52, P<0.001); however, fire too had a significant contribution (r2=0.23, P<0.001) to this axis. Using partitioning analysis, the combined effect of fire and sampling depth significantly explained 14% of the observed variation, whereas sample geochemistry explained 20% and interaction of all environmental parameters accounted for 22% (Figure 2b). Community composition was not significantly different at the surface when corrected for the loss of the organic horizon, whereas likely due to thawing of permafrost the impact of fire was more evident in the deeper layers (Figure 2c). The duplicate samples also clustered significantly more closely together in the thawed subsurface soils compared with the permafrost samples in the controls, suggesting that the communities in the thawed samples were more similar to each other compared with the permafrost. Significant differences in β-diversity were also correlated with differences in C and N content, followed by changes in sample pH and moisture (Figure 2c). Sample EC and DOC had smaller but significant contributions to the total variation.

16S rRNA gene sequencing with HiSeq2000 reveal the differences in prokaryotic diversity of samples from control and fire-impacted locations. (a) Communities clustered using principal coordinates analysis of the weighted UniFrac distance matrix. Each point corresponds to a sample colored in red/orange (○) for fire-impacted locations and in blue (◊) for the control locations. Lighter colors present the mineral soil depths. The percentage of variation explained by the plotted principal coordinates is indicated on the axes. (b) Contribution depth, fire and soil geochemistry on observed β-diversity was calculated via variation partitioning and represented as a Venn diagram. (c) Significant contributions of soil geochemistry to observed differences in prokaryotic diversity was tested using ANOVA.
The OTUs from all of the samples were distributed among 12 Bacterial and two Archaeal divisions, with the most abundant belonging to Acidobacteria, Actinobacteria and candidate division AD3 (Figure 3a). The most abundant phyla in the control surface layer samples were Actinobacteria, Verrucomicrobia, Acidobacteria, Alpha- and Gammaproteobacteria. At the control locations, there was a high abundance of Chloroflexi and AD3 at a depth of 10–20 cm. The middle layer in the control profiles had increasing abundances of Euryarchaeota, AD3 and Chloroflexi populations. Likewise, AD3 and Chloroflexi were highly abundant in the permafrost, whereas there was a decrease in representation of Proteobacteria and Acidobacteria with depth.

(a) Distribution of phylogenetic groups in the control and burned locations in each soil depth. (b) Pearson’s correlation tests were used to detect significant positive or negative correlations (yellow shading) of each phylum to the fire event or soil depth.
After the fire there were significant differences (R=0.42, P<0.001) in the composition of the surface layer community based on ANOSIM, with a reduction in relative abundances of Proteobacteria, Planctomycetes, Actinobacteria and Verrucomicrobia. By contrast, there was an average of a 68% increase in AD3 abundance after fire. At the 50–60 cm depth, Betaproteobacteria and Verrucomicrobia were significantly more abundant at fire-impacted locations. Finally in the deeper layers, Verrucomicrobia, Gemmatimonadetes and AD3 populations were higher in relative abundance, whereas Acidobacteria, Actinobacteria, Chloroflexi and all Proteobacteria had a significantly lesser relative abundance than in controls.
Comparisons between sampling strategy (burn × depth), soil geochemistry and OTU abundances (Figure 3b) showed that only AD3 and Gemmatimonadetes populations were positively and significantly correlated with the fire event. Acidobacteria and Proteobacteria abundances were not affected by fire, but were generally low in subsurface soils. Within Proteobacteria, Betaproteobacteria was an exception to this finding, where its abundances significantly increased with depth. In addition to depth and fire, other environmental parameters such as pH, C and N content were mainly correlated with AD3, Bacteroidetes, Gemmatimonadetes, Verrucomicrobia and Proteobacteria populations (Supplementary Table S4). In contrast, we did not observe any correlation between the soil geochemistry and the Archaeal community structure. Moreover, the total enzyme activity was positively correlated with Bacteroidetes, Gammaproteobacteria and Proteobacteria and negatively correlated with AD3 abundances. This could imply that the substrates used were not applicable to AD3 as its physiology is unknown.
Functional differences between control and fire-impacted microbial communities
Analysis of metagenomes from different soil depths showed that the fire event resulted in significant changes in functional gene content for the overall soil profile. As the organic horizon depth in burned locations were highly variable (11±9 cm) and 13C-NMR analysis suggested high microbial activity below the soil surface, we analyzed functional gene content in the depth of 10–20 cm. We used a de novo comparative metagenomic approach, namely Compareads (Maillet et al., 2012), to identify reads that were shared between paired metagenomes from different soil depths and locations. This approach enabled us, without prior annotation, to calculate pairwise similarities between unassembled short reads of the metagenomes and to classify differences in DNA contents between samples. The analysis revealed that in the control locations the metagenomes from each depth had high similarity to each other: 25.33% surface layers, 25.64% middle layers and 46.57% permafrost layers (Figure 4a). The metagenomes clustered within each sampling location despite the differences in sampling depths. In addition, the surface layer metagenomes from the control sites clustered separately not only from the middle layer and permafrost metagenomes but also from all the other metagenomes of fire-impacted locations. Intact middle and permafrost layers from the control sites showed high similarity to each other (26.24–28.56%). In contrast, in fire-impacted locations, metagenomes were less similar to each other than the ones in control locations: 1.67% surface soils, 22.22% middle and 25.99% deep soils.

(a) Heatmap of intersections in Nome Creek metagenomes. Similarity matrix resulting from the comparison of 11 samples using Compareads (khmer=33, t=2, 5.7E7 reads). Gray levels correspond to similarity levels. The three main groups, in blue, turquoise-blue and red, correspond, respectively, to control surface layer (S), control middle (M) and permafrost (D) and all layers from fire-impacted locations. Legend shows the fraction of similarity between the comparisons. (b) Between-class analysis, which visualizes results from PCA and clustering of the KO annotations from 12 metagenomes. The between-class analysis finds the principal components based on the center of gravity of each group as a result every symbol represent two replicate metagenomes. Two principal components are plotted using the ade4 package in R where (◊) surface, (Δ) middle and (○) permafrost (deep soil) layers. Blue color indicates control samples, whereas red indicates fire-impacted samples.
The metagenomic reads were screened using the FOAM database to determine which functional genes were present. Out of 617 KOs in the database, 486 KOs were detected in all metagenomes. This screening resulted in annotation of an average 432 416±95 402 genes per sample (Supplementary Table S3). Gene abundances (gene counts per KO) were normalized to the number of recombinase A copies detected in each metagenome. Both in control and fire-impacted locations, 95–97% of the genes originated from Bacterial species (Supplementary Figure S6). Archaeal and Eukaryotic (fungal) genes were considerably less abundant than Bacterial genes and burned sites had the lowest number of genes originating from Archaea and Fungi.
Overall functional differences between control and fire-impacted metagenomes were investigated via PCA (Figure 4b), with between-class analysis to identify the drivers of observed clustering. Two PCA axes could explain 63% of the observed variation and metagenomes of fire-impacted locations were significantly different (P<0.001) than control locations. In both control and burned sites, the surface soil metagenomes clustered separately from the rest. In the middle and deep layers, metagenomes from control and burned locations clustered away from each other; however, this clustering was not significantly impacted by fire (F=2.87, P=0.08). Clustering of the metagenomes was significantly correlated with the changes in environmental conditions, where pH (F=7.39, P=0.01) and moisture (F=2.49, P=0.05) content were the main drivers.
There were relatively few significant changes in genes involved in the C cycle when comparing the burned and control locations. Relative abundances of genes coding for carbohydrate metabolism and carbon fixation were only marginally different between the sampling depths (Figure 5 and Supplementary Figure S7). Only genes involved in galactose metabolism were significantly (F=9.404, P=0.022) less abundant in the middle and deeper soils after the fire. Multivariate statistical analysis did not reveal any significant correlation between individual KO and fire or environmental variables, which could be expected based on the complexity of carbon-processing pathways, many of which are not well characterized for uncultivated soil microbes. We compared the observed changes in relative gene abundances for four hydrolytic enzymes that decreased after fire: α-glucosidase, BG, CBH and XYL (Supplementary Figure S8) to measured potential enzyme activities at 4 °C. Of these, significant positive correlations were only found for XYL (Spearman’s ρ=0.943, P-value=0.016) activity.

Heat maps indicating differences in relative abundances of functional genes involved in carbon and nitrogen cycle in the Nome Creek metagenomes. Impact of the fire and sampling depth on the observed variation of the relative gene abundances was tested ANOVA analysis, where significant (P<0.05) factors are represented within parentheses.
On the basis of the relatively low abundance of genes in the metagenomes for methanogenesis, there was a low potential for CH4 production in the surface soils in general at the Nome Creek site, although the potential was significantly higher in the control locations (Figure 5 and Supplementary Figure S9). After fire there was a significant decrease in most of the genes for methanogenesis in the surface soils. In the deeper layers, there were more genes for the aceticlastic (F=13.946, P=0.009) pathways compared with the hydrogenotrophic pathway after fire.
The most abundant genes for the N cycle in the metagenomes were those for assimilation/mineralization pathways (Figure 5 and Supplementary Figure S10). Genes responsible for nitrogen assimilation were 54–88% more abundant in the burned subsurface soils. For example, glutamate dehydrogenase and synthetase gene relative abundances were 5–10% higher in fire-impacted middle and permafrost layers, indicating the potential for ammonia assimilation into amino acids. Moreover, the relative abundance of ammonia-generating nitrite reductase (nrfA) was 70% higher (P=0.043) in fire-impacted surface soils. Genes for denitrification and nitrate assimilation were present in all metagenomes, but had a higher representation (32–66%) in fire-impacted locations. However, in the surface soils, the relative abundance of nitrate reduction genes (narGH) were significantly (P=0.013) reduced by fire. Conversely, narGH genes were abundant in fire-impacted deeper soils. Genes responsible for assimilatory nitrate reduction were abundant at burned locations; in particular, ferredoxin-nitrite reductase (nir) significantly increased in the middle (P=0.012) as well as in the deeper (P=0.005) soils after the fire. Nitrite (NO2) and nitrous oxide (NO) reductases significantly decreased (16–32%) at the burned locations, suggesting the potential for accumulation of N2O. Relative abundances for genes involved in nitrogen fixation and nitrification were not significantly impacted by fire.
Genes for dissimilatory sulfite reductase (dsr), the sulfur oxidation (sox) gene complex mediating thiosulfate oxidation and the adenosine 5′-phosphosulfate (APS) reductase (apr) were present in all of the metagenomes (Supplementary Figure S11); however, genes coding for reductive pathways were relatively more abundant than those for the oxidative pathways. Genes for sulfate reduction were significantly (F=10.219, P=0.004) higher in the fire-impacted deep soil layers compared with the permafrost. Interestingly, the relative gene abundances of stress response genes did not change significantly between fire-impacted and control locations. Heat-shock, cold-shock, osmotic and salt adaptation genes (Supplementary Figure S12) had similar relative abundances across all metagenomes.
Discussion
Boreal fires alter the structure and function of ecosystems and thawing of permafrost due to increased surface soil temperatures, particularly in rocky upland conditions (Shenoy et al., 2011; Nossov et al., 2013). A recent study of the Nome Creek region suggested that the permafrost had undergone repeated episodes of fire-mediated degradation during the late Holocene (Jorgenson et al., 2013). In upland black spruce forests with coarse mineral soils and significant slope, rapid permafrost thaw and drainage of permafrost water consequently results in drier soils and alters biogeochemical cycling processes (Hart et al., 2005; Wang et al., 2012; Jorgenson et al., 2013). The extensive Boundary Fire surrounding Nome Creek, AK caused losses of 1.5–4.6 kg C m−2 and 0.03–0.14 kg N m−2, which is substantially higher than an average fire event in this region (Boby et al., 2010; Turetsky et al., 2010). The fire at Nome Creek resulted in burning of up to 25–30 cm of surface organic material. We found that 7 years after the fire, these soils still had significantly less C, nutrient and moisture contents up to 1 m depth. These factors, in addition to the increased mean soil temperatures (Johnstone et al., 2010; Nossov et al., 2013), could limit re-establishment of the original soil microbial communities and explain the significant differences in microbial community structure that we observed here.
Wildfires can result in immediate increased GHG fluxes to the atmosphere, mainly as CO2, resulting in a direct feedback to climate (Rustad et al., 2001). To understand the potential microbiological feedbacks on a longer time scale, we studied GHG production from different soil layers 7 years after the Boundary fire (Figure 1 and Supplementary Figure S2) and compared that with the predicted metabolic potential based on the metagenome data. We found that the CO2 production potential, based on gas flux measurements, was significantly reduced in the soils that had been impacted by the fire. In addition, the gas flux data revealed that the potential for CH4 and N2O production were negligible in all samples. The loss of readily degradable C and a corresponding decrease in moisture content (O’Donnell et al., 2009) are the likely explanations for the limited decomposition of remaining organic material in burned locations. The occurrence of buried cryoturbated organic masses in the soil, indicative of previous permafrost degradation events, also indicates buried organic matter can be persistent due to limited decomposition (Nossov et al., 2013).
We further investigated the state of the remaining surface organic matter using solid-state 13C-NMR. The composition of SOM was modified by fire, especially in the surface 0–10 cm of the burned soils. 13C-NMR analysis showed that in the post-fire soils microbial decomposition of C residues continued without fresh inputs below the first 10 cm (i.e. at 10–30 cm depths), as indicated by SOM with higher alkyl/O-alkyl ratios, characteristic of more decomposed organic material (Baldock et al., 1997; Norris et al., 2011). In accordance with previous reports of increasing alkyl/O-alkyl ratio with increasing depth in soil profiles (Baldock et al., 1997; Quideau et al., 2000), our results suggest the preferential decomposition of labile C sources such as carbohydrates and preservation of hydrophobic alkyl C pools (Baldock et al., 2004) in the soils.
The potential enzyme activity of soils can be interpreted both as an indicator of microbial activity and of substrate availability (Sinsabaugh et al., 2008). Our enzyme activity measurements revealed that the surface soils had higher potential enzymatic activities against a variety of substrates when compared with subsurface soils (Supplementary Figure S3) at both burned and control locations. However, when normalized to the total C content of the soils, the deeper soil layers had similar enzyme activities as the surface soils (Figure 1c). Therefore, it can be hypothesized that the subsurface soils had similar potential activity to surface soils, but were C limited. Higher enzymatic activities in active layer soils (compared with permafrost) were previously reported and are in agreement with our findings (Wallenstein et al., 2009; Waldrop et al., 2010; Coolen et al., 2011). Postfire, there was a marginal but significant increase in the potential enzymatic activities of carbohydrate-degrading enzymes, suggesting that despite the large reduction in C content, the potential microbial activity for degradation of carbohydrates was not reduced, similar to what was suggested by the 13C-NMR analysis for surface soils.
In this study, we found that the fire resulted in a significant change in the microbial β-diversity and community structure throughout the soil profile. Soil microbial diversity in the Arctic is highly variable (Neufeld and Mohn, 2005; Steven et al., 2008) and the microbial diversity in permafrost is generally lower than active layer soils (Gilichinsky et al., 2007; Steven et al., 2007; Yergeau et al., 2010; Mackelprang et al., 2011). In line with these findings, in control locations, we also detected higher α-diversity in active layer soils compared with deeper soil layers (Supplementary Figure S5). However, the microbial community structure was significantly different between mineral layer soils (Figure 2a) of burned and control locations.
Fire and fire-mediated changes in the soil geochemistry were strong drivers of the observed microbial community shifts, especially in deep soils (Figure 2). The most abundant phyla in Nome Creek samples, in general, were Verrucomicrobia, Chloroflexi and candidate phylum AD3. Both Verrucomicrobia and Chloroflexi populations were negatively affected by fire. Interestingly, candidate phylum AD3 was significantly positively impacted by the fire and was one of the most abundant community members in the thawed deeper layers postfire. The abundance of this phylum was also correlated to higher pH and decreasing C, nitrogen and moisture content. One hypothesis is that AD3 has specific capabilities that enable the cells to survive after fire in subsurface soils. Unfortunately, AD3 currently has no isolated representatives and its genomic and physiological properties are unknown. Previous 16S rRNA gene-based studies show that AD3 is a globally distributed, but locally rare phylum that is mainly observed in alpine and subalpine soil and sediments (Zhou et al., 2003; Costello, 2007; Nemergut et al., 2008). It had been previously postulated that AD3 was relatively more abundant in subsurface soils (up to 20 cm in depth), where nutrients and C content are low (Costello, 2007). Therefore, the deeper C-depleted postfire soils may have presented a favorable niche for members of this phylum.
In surface soils, wildfires have previously been reported to result in changes in soil bacterial diversity and community structure (Ferrenberg et al., 2013). Proteobacteria, Firmicutes, Chloroflexi, Acidobacteria, Actinobacteria and Bacteroidetes are often detected as the dominant phyla in Arctic soils (Gilichinsky et al., 2007; Steven et al., 2007, 2009; Yergeau et al., 2010; Wilhelm et al., 2011). Among these phyla, Proteobacteria, Firmicutes, Chloroflexi, Acidobacteria and Actinobacteria were also found to be abundant in the Nome Creek samples. In two previous studies, Firmicutes, Betaproteobacteria and Actinobacteria abundances were reported to increase postfire (Smith et al., 2008; Ferrenberg et al., 2013). However, in contrast with these findings, we found that Firmicutes and Actinobacteria abundances decreased after the fire, whereas Betaproteobacteria showed no significant correlation to the fire (Figure 2b). These differences could be due to the length of time between the fire and the sampling because the previous studies were of samples taken several days to 6 months after the fire occurred. In our case, we sampled 7 years after the fire to determine what pervasive impacts, if any, the fire had on the soil communities.
The lack of genomic information from uncultivated and understudied species hampers the detection and annotation of functional genes from metagenomes and introduces uncertainty to the comparisons made between samples and conditions. In the Nome Creek metagenome analysis, we aimed to overcome these challenges by combining annotation-dependent and -independent comparative metagenomics approaches. However, the fact that many functions from abundant phyla such as Verrucomicrobia, Chloroflexi and candidate phylum AD3 remain undiscovered should be taken into account when reviewing the metagenomics analysis.
Both de novo comparison of raw reads and PCA clustering of annotated genes showed that the fire-induced shift from a more moist, C-rich environment to a significantly drier and C-depleted state was correlated to significant changes in not only the phylogenetic profiles but also in the functional microbial gene profiles of Nome Creek samples (Figures 4a and b). PCA analysis of annotated gene relative abundances supported the observations from the Compareads analysis, which showed that the genomic potential was significantly different between surface soils of fire-impacted and control locations. However, close grouping of samples from fire-impacted middle and deeper layer soils in PCA analysis was not statistically significant. This analysis suggests that the extent of the similarity observed in PCA clustering could be an overestimation due to the analysis of data that is reduced to the genes that could be annotated. On the other hand, similar trends were observed in both analyses.
Shifts observed in the C- and nitrogen-cycling processes in the Nome Creek samples underline the fact that fire-mediated changes on the soil–physical parameters are strong drivers of microbial metabolic potential in upland boreal forests. In concurrence with the hypothesis that Arctic soils are nitrogen limited (Mack et al., 2004; Nordin et al., 2004), we too detected high C/N ratios and decreasing total nitrogen concentrations throughout the soil profile. After the fire, the nitrogen content was further reduced in all soil depths. The depletion of nitrogen after the fire could be as limiting for microbial growth and activity as the loss of C in this ecosystem.
When we screened the metagenomes for changes in genes involved in the C cycle, we found a significant increase in genes coding for galactose metabolism in surface soils (Figure 5, Supplementary Figures S8 and S9) and a reduction in genes for methanogenesis and methane oxidation (Figure 5 and Supplementary Figure S10). Both negligible CH4 fluxes and overall low relative methanogenesis gene abundances in control and fire-impacted soil profiles suggest that methanogenesis was not a major C-cycling process at this site. It had been previously shown that microbial communities respond rapidly to permafrost thaw in samples collected from organic lowland locations in Alaska (Mackelprang et al., 2011). These responses included increases in relative gene abundances for carbohydrate, (hemi)cellulose, chitin and sugar processing systems. In the case of Nome Creek, the slope resulted in drainage of available C and moisture after the fire, thus creating a sharp contrast to the lowland site used in the study by Mackelprang et al. (2011). For the N cycle, we observed significant increases in ammonia and nitrate assimilation and low NO to N2 production potentials. Also, there was negligible N2O flux at the site. Similar findings were reported for other intact permafrost and surface layer samples, where genes involved in nitrogen fixation and ammonia oxidation exhibited low diversity and abundance in the metagenomic libraries (Yergeau et al., 2010). These data suggest that most of the available nitrogen was assimilated into microbial biomass.
Conclusions
Wildfire frequency in the boreal forest of Alaska has more than doubled over the past century (Kasischke and Turetsky, 2006) and can accelerate permafrost degradation, particularly in rocky uplands (Johnstone et al., 2010). The wildfires studied here were part of the 2004/2005 wildfire season, which were the largest on record (Turetsky et al., 2010). In Nome Creek, fire resulted in permafrost thaw and a subsequent decrease in soil moisture due to subsurface drainage at the site. Seven years after the fire event, there remained significant changes in the microbial community composition, in particular an increase in candidate phylum AD3. The fire thus had a pervasive impact on the microbial community. The fire impact was also reflected in 13C-NMR analysis, GHG flux measurements and potential soil enzyme activities that indicated reduced microbial potential for decomposition of SOM after the fire. The deep sequencing metagenomics approach that we used here enabled us to gain insight into the impact of fire on the microbial metabolic potential and to present unique evidence that fire not only changes the soil C storage and microbial community structure but also affects functional pathways throughout the soil profile.
References
Allison SD, McGuire KL, Treseder KK . (2010). Resistance of microbial and soil properties to warming treatment seven years after boreal fire. Soil Biol Biochem 42: 1872–1878.
Baldock JA, Oades JM, Nelson PN, Skene TM, Golchin A, Clarke P . (1997). Assessing the extent of decomposition of natural organic materials using solid-state 13C-NMR spectroscopy. Soil Res 35: 1061–1084.
Baldock JA, Masiello CA, Gélinas Y, Hedges JI . (2004). Cycling and composition of organic matter in terrestrial and marine ecosystems. Mar Chem 92: 39–64.
Bergner B, Johnstone J, Treseder KK . (2004). Experimental warming and burn severity alter soil CO2 flux and soil functional groups in a recently burned boreal forest. Global Change Biol 10: 1996–2004.
Berhe AA, Suttle KB, Burton SD, Banfield JF . (2012). Contingency in the direction and mechanics of soil organic matter responses to increased rainfall. Plant Soil 358: 371–383.
Boby LA, Schuur EA, Mack MC, Verbyla D, Johnstone JF . (2010). Quantifying fire severity, carbon, and nitrogen emissions in Alaska's boreal forest. Ecol Appl 20: 1633–1647.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R et al. (2012). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10: 57–59.
Bond-Lamberty B, Wang C, Gower ST . (2004). Net primary production and net ecosystem production of a boreal black spruce wildfire chronosequence. Global Change Biol 10: 473–487.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Chessel D, Dufour A-B, Jombart JRL, Ollier S, Pavoine S, Thioulouse J et al. (2012). Package ‘ade4’: Analysis of Ecological Data: Exploratory and Euclidean Methods in Environmental Sciences. R-package Version: 1.5-1 Foundation for Statistical Computing: Vienna, Austria.
Coolen MJL, van de Giessen J, Zhu EY, Wuchter C . (2011). Bioavailability of soil organic matter and microbial community dynamics upon permafrost thaw. Environ Microbiol 13: 2299–2314.
Costello EK . (2007) Molecular Phylogenetic Characterization of High Altitude Soil Microbial Communities and Novel, Uncultivated Bacterial Lineages. ProQuest: Ann Arbor, MI, USA.
Czimczik CI, Trumbore SE, Carbone MS, Winston GC . (2006). Changing sources of soil respiration with time since fire in a boreal forest. Global Change Biol 12: 957–971.
Ferrenberg S, O'Neill SP, Knelman JE, Todd B, Duggan S, Bradley D et al. (2013). Changes in assembly processes in soil bacterial communities following a wildfire disturbance. ISME J 7: 1102–1111.
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE et al. (2010). The Pfam protein families database. Nucleic Acids Res 38: D211–D222.
Gilichinsky D, Wilson G, Friedmann E, McKay C, Sletten R, Rivkina E et al. (2007). Microbial populations in Antarctic permafrost: biodiversity, state, age, and implication for astrobiology. Astrobiology 7: 275–311.
Gordon A, Tallas M, van Cleve K . (1987). Soil incubations in polyethylene bags: effect of bag thickness and temperature on nitrogen transformations and CO2 permeability. Can J Soil Sci 67: 65–76.
Harden JW, Trumbore SE, Stocks BJ, Hirsch A, Gower ST, O'Neill KP et al. (2000). The role of fire in the boreal carbon budget. Global Change Biol 6: 174–184.
Hart SC, DeLuca TH, Newman GS, MacKenzie MD, Boyle SI . (2005). Post-fire vegetative dynamics as drivers of microbial community structure and function in forest soils. Forest Ecol Manage 220: 166–184.
Hyatt D, Chen G-L, LoCascio P, Land M, Larimer F, Hauser L . (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform 11: 119.
Johnstone JF, Chapin FS, Hollingsworth TN, Mack MC, Romanovsky V, Turetsky M . (2010). Fire, climate change, and forest resilience in interior Alaska. Can J Forest Res 40: 1302–1312.
Jorgenson MT, Romanovsky V, Harden J, Shur Y, O’Donnell J, Schuur EAG et al. (2010). Resilience and vulnerability of permafrost to climate change. This article is one of a selection of papers from The Dynamics of Change in Alaska’s Boreal Forests: Resilience and Vulnerability in Response to Climate Warming. Can J Forest Res 40: 1219–1236.
Jorgenson MT, Harden J, Kanevskiy M, O’Donnell J, Wickland K, Ewing S et al. (2013). Reorganization of vegetation, hydrology and soil carbon after permafrost degradation across heterogeneous boreal landscapes. Environ Res Lett 8: 035017.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484.
Kasischke ES, Turetsky MR . (2006). Recent changes in the fire regime across the North American boreal region—spatial and temporal patterns of burning across Canada and Alaska. Geophys Res Lett 33: L09703–L09708.
Kelsey KC, Wickland KP, Striegl RG, Neff JC . (2012). Variation in soil carbon dioxide efflux at two spatial scales in a topographically complex boreal forest. Arct Antarct Alp Res 44: 457–468.
Legendre P, Gallagher ED . (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129: 271–280.
Lehmann J, Czimczik C, Laird D, Sohi S . (2009). Stability of biochar in soil. In: Lehmann J, (ed). Biochar for Environmental Management: Science and Technology. Earthscan: London, pp 183–205.
Mack MC, Schuur EAG, Bret-Harte MS, Shaver GR, Chapin FS . (2004). Ecosystem carbon storage in arctic tundra reduced by long-term nutrient fertilization. Nature 431: 440–443.
Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480: 368–371.
Maillet N, Lemaitre C, Chikhi R, Lavenier D, Peterlongo P . (2012). Compareads: comparing huge metagenomic experiments. BMC Bioinform 13: S10.
McGuire AD, Chapin FS III, Wirth C, Apps M, Bhatti J, Callaghan T et al. (2007). Responses of high latitude ecosystems to global change: potential consequences for the climate system. In: Canadell J, Pataki D, Pitelka L, (eds) Terrestrial Ecosystems in a Changing World. Springer: Berlin, Germany, pp 297–310.
Nemergut DR, Townsend AR, Sattin SR, Freeman KR, Fierer N, Neff JC et al. (2008). The effects of chronic nitrogen fertilization on alpine tundra soil microbial communities: implications for carbon and nitrogen cycling. Environ Microbiol 10: 3093–3105.
Neufeld JD, Mohn WW . (2005). Unexpectedly high bacterial diversity in arctic tundra relative to boreal forest soils, revealed by serial analysis of ribosomal sequence tags. Appl Environ Microbiol 71: 5710–5718.
Nordin A, Schmidt IK, Shaver GR . (2004). Nitrogen uptake by Arctic soil microbes and plants in relation to soil nitrogen supply. Ecology 85: 955–962.
Norris CE, Quideau SA, Bhatti JS, Wasylishen RE . (2011). Soil carbon stabilization in jack pine stands along the boreal forest transect case study. Global Change Biol 17: 480–494.
Nossov DR, Jorgenson MT, Kielland K, Kanevskiy MZ . (2013). Edaphic and microclimatic controls over permafrost response to fire in interior Alaska. Environ Res Lett 8: 035013.
O'Donnell JA, Harden JW, McGuire AD, Kanevskiy MZ, Jorgenson MT, Xu X . (2011). The effect of fire and permafrost interactions on soil carbon accumulation in an upland black spruce ecosystem of interior Alaska: implications for post-thaw carbon loss. Global Change Biol 17: 1461–1474.
O’Donnell J, Turetsky M, Harden J, Manies K, Pruett L, Shetler G et al. (2009). Interactive effects of fire, soil climate, and moss on CO2 fluxes in Black Spruce ecosystems of interior Alaska. Ecosystems 12: 57–72.
Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ et al. (2007) The Vegan Package. Community Ecology Packagehttp://www.r-project.org Vol. 10, 2008.
Osterkamp TE, Romanovsky VE . (1999). Evidence for warming and thawing of discontinuous permafrost in Alaska. Permafrost Periglac 10: 17–37.
Price MN, Dehal PS, Arkin AP . (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650.
Quideau S, Anderson M, Graham R, Chadwick O, Trumbore S . (2000). Soil organic matter processes: characterization by 13C NMR and 14C measurements. Forest Ecol Manage 138: 19–27.
Ramette A . (2007). Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62: 142–160.
Randerson JT, Liu H, Flanner MG, Chambers SD, Jin Y, Hess PG et al. (2006). The impact of boreal forest fire on climate warming. Science 314: 1130–1132.
Rustad L, Campbell J, Marion G, Norby R, Mitchell M, Hartley A et al. (2001). A meta-analysis of the response of soil respiration, net nitrogen mineralization, and aboveground plant growth to experimental ecosystem warming. Oecologia 126: 543–562.
Schuur EAG, Bockheim J, Canadell JG, Euskirchen E, Field CB, Goryachkin SV et al. (2008). Vulnerability of permafrost carbon to climate change: implications for the global carbon cycle. BioSci 58: 701–714.
Shenoy A, Johnstone JF, Kasischke ES, Kielland K . (2011). Persistent effects of fire severity on early successional forests in interior Alaska. Forest Ecol Manage 261: 381–390.
Sinsabaugh RL, Lauber CL, Weintraub MN, Ahmed B, Allison SD, Crenshaw C et al. (2008). Stoichiometry of soil enzyme activity at global scale. Ecol Lett 11: 1252–1264.
Smith NR, Kishchuk BE, Mohn WW . (2008). Effects of wildfire and harvest disturbances on forest soil bacterial communities. Appl Environ Microbiol 74: 216–224.
Steven B, Briggs G, McKay CP, Pollard WH, Greer CW, Whyte LG . (2007). Characterization of the microbial diversity in a permafrost sample from the Canadian high Arctic using culture-dependent and culture-independent methods. FEMS Microbiol Ecol 59: 513–523.
Steven B, Pollard WH, Greer CW, Whyte LG . (2008). Microbial diversity and activity through a permafrost/ground ice core profile from the Canadian high Arctic. Environ Microbiol 10: 3388–3403.
Steven B, Niederberger TD, Whyte LG . (2009). Bacterial and archaeal diversity in permafrost. In: Margesin R (ed). Permafrost Soils. Springer: Berlin, Germany, pp 59–72.
Tarnocai C, Canadell JG, Schuur EAG, Kuhry P, Mazhitova G, Zimov S . (2009). Soil organic carbon pools in the northern circumpolar permafrost region. Global Biogeochem Cycle 23: GB2023.
Turetsky MR, Kane ES, Harden JW, Ottmar RD, Manies KL, Hoy E et al. (2010). Recent acceleration of biomass burning and carbon losses in Alaskan forests and peatlands. Nat Geosci 4: 27–31.
Turetsky MR, Donahue WF, Benscoter BW . (2011). Experimental drying intensifies burning and carbon losses in a northern peatland. Nat Commun 2: 514.
Waldrop MP, Harden JW . (2008). Interactive effects of wildfire and permafrost on microbial communities and soil processes in an Alaskan black spruce forest. Global Change Biol 14: 2591–2602.
Waldrop MP, Wickland KP, White Iii R, Berhe AA, Harden JW, Romanovsky VE . (2010). Molecular investigations into a globally important carbon pool: permafrost-protected carbon in Alaskan soils. Global Change Biol 16: 2543–2554.
Wallenstein MD, McMahon SK, Schimel JP . (2009). Seasonal variation in enzyme activities and temperature sensitivities in Arctic tundra soils. Global Change Biol 15: 1631–1639.
Wang G, Garcia D, Liu Y, de Jeu R, Johannes DA . (2012). A three-dimensional gap filling method for large geophysical datasets: Application to global satellite soil moisture observations. Environ Modell Softw 30: 139–142.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wilhelm RC, Niederberger TD, Greer C, Whyte LG . (2011). Microbial diversity of active layer and permafrost in an acidic wetland from the Canadian High Arctic. Can J Microbiol 57: 303–315.
Yergeau E, Hogues H, Whyte LG, Greer CW . (2010). The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J 4: 1206–1214.
Zhou J, Xia B, Huang H, Treves DS, Hauser LJ, Mural RJ et al. (2003). Bacterial phylogenetic diversity and a novel candidate division of two humid region, sandy surface soils. Soil Biol Biochem 35: 915–924.
Acknowledgements
This work was supported in part by the Director, Office of Science, Office of Biological and Environmental Research, Climate and Environmental Science Division, of the US Department of Energy under Contract No. DE-AC02-05CH11231 as part of the Terrestrial Ecosystem Science Program to Lawrence Berkeley National Laboratory, the Keck Foundation (Earth Microbiome Project; http://www.earthmicrobiome.org) and the United States Geological Survey (USGS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Taş, N., Prestat, E., McFarland, J. et al. Impact of fire on active layer and permafrost microbial communities and metagenomes in an upland Alaskan boreal forest. ISME J 8, 1904–1919 (2014). https://doi.org/10.1038/ismej.2014.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.36
Keywords
This article is cited by
-
Elevated methane flux in a tropical peatland post-fire is linked to depth-dependent changes in peat microbiome assembly
npj Biofilms and Microbiomes (2024)
-
Distinct taxonomic and functional profiles of high Arctic and alpine permafrost-affected soil microbiomes
Environmental Microbiome (2023)
-
Soil viral diversity, ecology and climate change
Nature Reviews Microbiology (2023)
-
Permafrost microbial communities and functional genes are structured by latitudinal and soil geochemical gradients
The ISME Journal (2023)
-
Post-fire soil emissions of nitric oxide (NO) and nitrous oxide (N2O) across global ecosystems: a review
Biogeochemistry (2023)
