
Abstract
The human gastrointestinal (GI) microbiota is important to human health and imbalances or shifts in the gut microbial community have been linked to many diseases. Most studies of the GI microbiota only capture snapshots of this dynamic community at one or a few time points. Although this is valuable in terms of providing knowledge of community composition and variability between individuals, it does not provide the foundation for going beyond descriptive studies and toward truly predictive ecological models. In order to achieve this goal, we need longitudinal data of appropriate temporal and taxonomic resolution, so that established time series analysis tools for identifying and quantifying putative interactions among community members can be used. Here, we present new analyses of existing data to illustrate the potential usefulness of this approach. We discuss challenges related to sampling and data processing, as well as analytical approaches and considerations for future studies of the GI microbiota and other complex microbial systems.
Similar content being viewed by others


Introduction
Statistical analysis of time series is an established tool in ecology for purposes like forecasting (Ferrari et al., 2008), delineation of ecosystem stability properties (Turchin and Taylor, 1992) and estimation of interactions between community members (Stenseth et al., 1997). Importantly, time series analysis can provide insight into the factors that determine temporal demographic fluctuations, that is, population and community dynamics, in ecological systems and thus provide information on how communities of organisms will respond to perturbations. This type of knowledge is essential for successful ecosystem management, yet this approach has been somewhat underused in microbiology. This does not necessarily stem from microbial systems being fundamentally different from plant and animal systems in terms of community ecology, but rather a tradition of reductionism in microbiology, as well as previous difficulties in reliably measuring demographic variables in complex microbial systems. However, the advent of high-throughput DNA-based methods for assessing microbial community composition in complex samples has helped microbiology become more systems oriented, and detailed time-resolved surveys are now highly feasible. In this perspective article, we demonstrate the power of a time series approach for studying the ecology of the gastrointestinal (GI) microbiota and we argue that this and similar approaches should be considered when designing future studies.
The animal gut is the most densely populated ecosystem known to science with the microbial cells residing in the gut of a healthy human outnumbering host cells by more than an order of magnitude (Sommer and Backhed, 2013). Presently, we have a fairly nuanced view of taxonomic and metabolic diversity of the GI microbiota (Human Microbiome Project C, 2012). However, the realization that this complex community of microbes forms an integral part of the human organism (Cho and Blaser, 2012) has prompted calls for a more profound understanding that transcends descriptive studies (Greenblum et al., 2013). Lamentably, there is still a relative paucity of large-scale longitudinal studies that allow us to observe how populations of the various members of the GI microbial community fluctuate over time, and explore drivers of ecosystem dynamics. The GI microbial community is heavily influenced by host factors that make up the gut environment such as diet (David et al., 2014), immune function (Hooper et al., 2012) or drug use (Dethlefsen and Relman, 2011). These and other factors constitute the parameter space within which interactions between groups of microorganisms occur (Trosvik et al., 2010).
There are three main approaches to inferring biotic interactions:
Competition experiments: biotic interactions can be inferred by analysing time series data from controlled competition experiments in the laboratory (Faust and Raes, 2012). In the case of the GI microbiota, this is, in general, not a viable option as most species are non-cultivable (Zoetendal et al., 2004). Also, the form of a pairwise interaction may depend upon the environmental context in which it takes place (de Muinck et al., 2013), making extrapolation from laboratory experiments to natural systems problematic.
Co-occurrence modelling: this approach is sometimes used in cross-sectional community studies (Faust and Raes, 2012), and is particularly valuable for investigating environmental effects. Inference of biotic interactions from this type of data is based on the rationale that negatively correlated occurrence patterns arise from negative interactions, like competition, whereas positive correlation patterns stem from positive interactions such as cross-feeding. Although not unreasonable, these are quite strong assumptions and their general validity is debated (Faust and Raes, 2012) as correlation is neither necessary nor sufficient to establish causation and may result from groups of microbes having either similar or opposing responses to environmental factors. This sort of concerted behaviour in the GI microbiota has been documented by Gerber et al. (2012) who devised an algorithm for identification of coordinated responses across taxa after perturbations. The inference problem is partially related to the concept known as Granger causality, which states that two variables can be said to be causally related if a lagged form of one variable enhances predictability of the other (Granger, 1969). Granger causality is recognized as the primary advance on the causation problem since Berkley in the early seventeenth century (Sugihara et al., 2012), and cannot be inferred from cross-sectional data, which compares physically separated ecosystems.
Community time series analysis: an approach based on observations of changing population abundances within a single system over time is needed in order to say with confidence that one population is affecting the dynamics of another. The main purpose of this perspective article is to advocate that statistical analysis of comprehensive longitudinal studies of high temporal resolution constitutes an invaluable means of describing biotic interaction structures, and thus form an essential link between descriptive studies and a predictive systems-level framework for the GI microbiota.
Longitudinal studies of the human gut microbiota
A handful of longitudinal studies have explored the human GI microbiota with various scopes and methodologies, in all cases demonstrating a highly dynamic community. The premature infant gut has been found to host a low diversity community that develops through a succession of aerobic and facultative aerobic bacteria (Morowitz et al., 2011; Sharon et al., 2013). Full-term infants are usually dominated by Proteobacteria and Firmicutes early on with abundances of Bacteroidetes increasing with age, and the community converging on an adult-like profile dominated by Bacteroides and Firmicutes within 1–2 years (Palmer et al., 2007; Koenig et al., 2011). Longitudinal studies of the adult GI microbiota have shown that it responds rapidly to perturbations like antibiotic treatment (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011) and dietary change (David et al., 2014), but that the community generally recovers upon cessation of treatment or return to a regular diet. Long-term studies have shown that although population abundances in the gut community fluctuate substantially over time, many species are retained for decades (Rajilic-Stojanovic et al., 2012; Faith et al., 2013). All of the studies listed above are characterized by a trade-off between duration and temporal resolution making application of typical time series analysis techniques difficult. In contrast, Caporaso et al. (2011) followed one male and one female subject with samples collected frequently, over a period of 443 days (332 samples) and 185 days (130 samples), respectively (Table 1). Owing to the exceptional temporal scope and resolution of this study, we will use the data to illustrate our main points, with a particular focus on the longest (male) series.
Materials and methods
The data analysed here are the operational taxonomic unit (OTU) tables made available as Supplementary material in (Caporaso et al., 2011). Briefly, ribosomal RNA gene V4 fragments were sequenced using Illumina GAIIx and 97% OTUs were assigned from the Greengenes database (DeSantis et al., 2006) using uclust (Edgar, 2010). For details, see (Caporaso et al., 2011).
All statistical analyses were done with R (R Core Team, 2014) with the following non-standard R packages used for specific methods: The car package was used for Levene’s tests for homogeneity of variance using medians as group centres. Vegan was used for OTU subsampling to the lowest number of sequence reads (Table 1) without replacement. Mgcv was used for fitting GAMs using penalized regression splines with three degrees of freedom for defining smooth terms for independent variables. Network was used for network visualization with default settings, based on a binarization of the matrix of P-values from the all vs all time-lagged linear regression analysis (that is, P⩽0.01 was coded as 1, while all other cells were coded as 0).
Phenomenological classification of GI bacterial population states
When looking at the Caporaso data in detail, certain phenomena that we would not be able to observe in cross-sectional studies emerge, underlining the importance of high-resolution time series studies for establishing a benchmark for normal temporal variation in the GI microbiota.
Stability of GI bacterial populations
Different GI bacterial taxa have different propensities for abundance fluctuation (Figure 1a). For both subjects, roughly half of the genera that are detected at least twice vary in abundance by less than one order of magnitude. Conversely, a handful of genera, including Escherichia, Akkermansia and Prevotella, vary by as much as four orders of magnitude. Interestingly, the same OTUs tend to be highly dynamic or relatively stable in the two subjects (rho=0.72, P≪0.001, Spearman’s rank correlation). This kind of information can only be gleaned through longitudinal studies, and is useful for interpretation of cross-sectional data. Observing this type of behaviour is highly contingent on the taxonomic scale of observation (Box 1). For instance, the most dominant phylum, the Bacteroidetes, is relatively stable over time, fluctuating by considerably less than one order of magnitude. By shifting the taxonomic scale of observation to the level of the order (Bacteroidales), it becomes apparent that this group is made up of both highly variable and relatively stable genera (Figure 1b). This is difficult to explain, but it may be that relative stability on a broad taxonomic scale is caused by direct or indirect host modulation acting on high taxonomic levels because of functional redundancies among different taxon members.

Dynamic patterns in the healthy adult human gut. (a) Dynamic range of observed genera measured as the log ratio of the highest to the lowest abundances of genera found in at least two samples per subject (y axis) rank ordered from lowest to highest dynamic range (x axis). The rank orderings of the 147 genera that are observed at least twice in both individuals are highly correlated (rho=0.72, P≪0.001, Spearman’s rank correlation). (b) Dynamic patterns within the order Bacteroidales are illustrated by the relative stability of some genera and abrupt fluctuations in the abundances of others. Stable taxa include Bacteroidales*, Bacteroides, Parabacteroides, Alistipes. Fluctuating taxa include Prevotella and Porphyromonas. Besides qualitative assessment of the graph, the ratio of the mean to the s.d. was used as a rough metric to support this classification. (c) Regime shift within the phylum Proteobacteria. The Proteobacteria are dominated by the gamma, delta and epsilon classes until around day 100 when the beta class becomes dominant. The small dark blue spikes at the bottom of the plot are Alphaproteobacteria. (d) Transiently dominant classes within the phylum Tenericutes are observed as blooms. Erysipelotrichi are partially displaced by ML615J-28 and then Mollicutes. Data in (b–d) are from the male subject with the taxonomic group names indicated in the colour keys above the plots. *Classified to order level.
In addition to the general patterns described above, we find two related categories of temporal demographic patterns that entail extensive re-modelling of population structure. These phenomena, which we term ‘Regime shifts’ and ‘Transients’, are described below.
Regime shifts
In biology, regime shifts are defined as large, abrupt and long-lasting changes in the structure of an ecosystem (Biggs et al., 2009). This class of phenomena is of particular interest as abrupt and persistent changes in GI microbiota structure can have important phenotypic effects on the host (Clemente et al., 2012). When focusing on the intra-taxon dynamics of Proteobacteria in the male subject (Figure 1c), we observe an initial subcommunity dominated by Deltaproteobacteria, Gammaproteobacteria and Epsilonproteobacteria. Around day 100, there is an abrupt and persistent switch to a community structure characterized by high abundances of Betaproteobacteria. Remarkably, a very similar shift is seen in the female subject, although in this case the subcommunity at the start of the observation period is largely devoid of Epsilonproteobacteria and the regime shift occurs around day 50. Reliable detection of impending regime shifts is difficult but analysis of ecological time series can provide indicators like increased variability and autocorrelation (Biggs et al., 2009).
Transients
A related but distinct phenomenon occurs when abrupt changes in community structure are not persistent and thus cannot be classified as regime shifts, but rather as transient sustained outgrowths. Initially in the male subject, we observe a single class of the phylum Tenericutes, Erysipelotrichi (Figure 1d). Within this background, two temporally separated blooms can be observed. The first involving a taxon designated ML615J-28 and the second belonging to the class Mollicutes. We cannot know for certain whether the transient groups were introduced from an external source or that they were not observed in a majority of samples because of insufficient sampling. However, the fact that these groups are observed in a large number of consecutive time points strongly suggests that the outgrowths are not merely sampling artefacts.
Sampling probabilities and classification bias
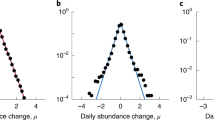
An early step in inferring biotic interactions from time series data are to accurately characterize variations in community composition from one time point to another. There are several sources of bias that need to be considered when interpreting metagenomic community survey data, although these are not exclusive to longitudinal studies. Assuming a total bacterial density of 1011 cells per gram of faeces, consider a population of 105 cells. With a sampling depth of 105 reads, there would still be a probability of <0.1 of sampling a single cell, and for a population of 104 cells one would have to sample millions of sequences in order to have a good chance of observing a single read from that population. Clearly, we are more likely to sample rare populations at higher sampling depths, and bias introduced by variable sampling effort is usually addressed by subsampling to an even sequencing depth (Figure 2a).

Sampling effort, rare populations and diversity in the Caporaso data. (a) Observed number of genus-level OTUs plotted against number of reads per sample with data pooled from both subjects for raw data (black) and data subsampled to the lowest number of reads (red). The lines are linear regression fits. Without subsampling, there is a highly significant relationship between (R2=0.14, P≪0.001) between observed richness and sampling depth. Subsampling makes the association weaker (R2=0.03), but the relationship is still significant (P<0.001). (b) Distribution of the number of genera-level OTUs in the male subject that are found in at least the number of samples indicated on the x axis for raw data (black) and data subsampled to the lowest number of reads (red). For instance, using the raw data 38 OTUs were observed in at least 90% of the samples, 64 were observed in at least 50% of the samples, whereas 107 OTUs were observed in at least 10% of samples. For subsampled data, the corresponding numbers were 30, 53 and 91 OTUs, respectively. (c) Shannon entropy over time, as measured for genus-level OTUs, for the male (black) and female (red) subject. The dotted lines are means. The mean and the variance are elevated in the male relative to the female (P≪0.001, t-test and Levene’s test, respectively). (d) Relationships between specific phylum abundances and Shannon entropy for genus-level OTUs. The green (Bacteroidetes) and blue (Firmicutes) lines are linear regression fits (R2=0.81 and 0.72, respectively, P≪0.001 in both cases). Turquoise (Actinobacteria) and pink (Proteobacteria) lines are smoothing splines fitted with three degrees of freedom (R2=0.74 and 0.07, respectively, P≪0.001 in both cases, GAMs).
In the Caporaso data, most genera appear as rare as they are observed only in very few samples (Figure 2b). For instance, more than two-thirds of the genera observed in the male are found in <10% of the samples, and more than one-fourth are observed in only one. This pattern remains similar after subsampling to the lowest number of reads. It is important to note that several technical factors are known to influence short read amplicon sequencing data, such as choice of primers (Soergel et al., 2012), library preparation method (van Dijk et al., 2014) and the algorithm and training taxonomy used for classification (Werner et al., 2012; Mizrahi-Man et al., 2013). These factors can bias the data toward OTUs that are more readily classified given the set of methods used. In the time series context, as long as biases are systematic, statistical analysis of community dynamics is still feasible, although one should still be cautious of misclassification. Several analytical approaches, like dynamic regression models, will however, be hampered by the fact that many OTUs are observed with a low probability, typically leading to sparse OTU matrices. These types of analyses are thus restricted to populations that can be reliably and consistently quantified.
Intra-individual temporal variation in community diversity
Understanding the effects of taxonomic diversity in the GI microbiota is important for linking ecological resilience and stability with clinically relevant community configurations (Lozupone et al., 2012). In the Caporoso data, Shannon entropy and the number of observed genus-level OTUs (richness) are extremely variable over time (Figure 2c, Supplementary Figures S1 and S2), and both diversity metrics are significantly correlated with sampling depth (Figure 2a and Supplementary Figure S3), although the relationship between richness and sampling effort is weakened by subsampling to the lowest sequencing depth. Therefore, all subsequent analyses presented in this section were carried out in parallel on raw (Figure 2, Supplementary Figures S2A, S3, S4A, S6 and S8) and subsampled (Figures 2a and b, Supplementary Figures S1, S2B, S3, S4B, S5, S7 and S9) data. It is noteworthy that the observed diversity is much higher in the male relative to the female (Figure 2c, Supplementary Figures S1 and S2). The variances in Shannon entropy also differ significantly between the two individuals (Figure 2c, Supplementary Figure S1), which can be attributed to increased variance in the evenness rather than richness in the male (Supplementary Figures S2 and S4). By looking at how different groups of bacteria are associated with variation in diversity indices over time (Figure 2d and Supplementary Figures S5–S9), we can get a general idea of what is underlying these observations. There is a strong negative correlation between abundances of Bacteroidetes and diversity, while the opposite trend is seen for Firmicutes, Actinobacteria and Proteobacteria. Note that the observed richness will be affected by the underlying abundance distribution (evenness) in a sample, for example, if one OTU is present at very high relative abundance in a sample (low evenness) there will be less sequencing effort dedicated to less abundant OTUs, thus negatively affecting the observed richness. Also, reliable classification was only possible to the genus level in this case and our observations may not provide an accurate reflection of diversity on lower taxonomic levels.
Biotic interaction networks
Time series regression models
A statistical test for identification and quantification of biotic interactions is regression of a variable describing the abundance dynamics of a taxon of interest on time-lagged abundances of another taxon. Thus, this approach provides stronger evidence for biotic interactions than correlated co-occurrence patterns. In order to capture the dynamics of a population in a single variable, one can use the per unit time difference in abundances, that is, xi,t+1–xi,t, where xi,t is the abundance of taxon i at time t (usually log transformed). Using an appropriate regression technique, this analysis will find a significant negative relationship between the dependent and independent variables if detectable competition is occurring. Conversely, one would observe a significant positive relationship if one organism is benefitting from the presence of another. In the case of a complex system like the GI microbiota, this entails solving a large number of equations (Figure 3a). Differences in sampling depth between two consecutive time points can be assumed to result from random processes and thus should not introduce spurious associations. However, using raw counts can lead to misrepresentation of the dependent variables, such as if an observed increase in the abundance of a population from time t to t+1 is due to increased sampling rather than population expansion. It is thus generally preferable to use relative abundances for regression models, but if variation in sampling effort is not too extreme, then the same analysis carried out using raw count data should produce qualitatively similar results.

Time series regression approach. (a) System of equations for evaluating biotic interactions assuming simple linear relationships: Δxi,t=xi,t+1–xi,t, where xi,t is the log relative abundance of taxon i at time=t. αi,j are intercept terms, βi,j are linear regression coefficients and xj,t are log relative abundances of taxon j at time=t. The total number of equations is equal to n2, where n is the total number of taxa. In the male time series, n=38 when using the stated filtering criteria. (b) Heat map describing the strength and direction (βi,j in a) of highly significant interactions between genera of bacteria in a healthy adult human gut (male subject), estimated by using linear regression on the equation set in (a). Dependent variables are along the y axis and independent variables along the x axis. The colour key on the right-hand side indicates the sign and magnitude of interactions that were significant at the 99% confidence level. Cells representing nonsignificant relationships are black. Axis labels are colour coded according to phylum provenance of the genera as in (c). (c) Network representation of the biotic interactions identified in the previous steps demonstrating varying degrees of connectedness of the different genera. The network is based on a binary version of (b) where interactions significant at P⩽0.01 (coloured cells) were coded as 1 and all others (black cells) were coded as 0.
A further issue in time series analysis is that of missing data. This can occur when sampling is incomplete or irregular. The data must be culled to remove samples that cannot be organized into regularly spaced pairs. Although sampling in the study by Caporaso et al. (2011) was mostly on a daily basis there are considerable discontinuities in the series (Table 1), but culling the male series still leaves 269 out of 332 data points. Another way that data can be missing are when taxa are not observed in samples because of limitations in sequencing depth. Regression analysis should only be carried out with taxa for which there are large numbers of non-zero data points in order to ensure statistical power and facilitate log transformation of the data. In the analysis presented here only taxa observed in at least 90% of samples were included (total of 38 genera) and all value pairs containing a zero were excluded from model computations.
The functional relationships that we estimate by regression need not always be linear. Therefore, regression techniques that are flexible regarding assumptions made about the specific form of the functional relationship are valuable for inferring interactions. One such technique is the generalized additive model (GAM), which is a regression method where non-parametric spline functions are fitted to the data and evaluated within a probabilistic framework (Hastie and Tibshirani, 1990). This entails replacing equations in Figure 3a with xi,t+1–xi,t=αi,j+si,j(xj,t), where sij represents a smoothing function. This technique can detect complex non-linear associations between variables without prior specification of a functional relationship. GAMs do not output regression coefficients for smooth terms, but they estimate approximate P-values and explained variance, and functional forms can be visualized by plotting. An excellent implementation of GAMs can be found in the R package mgcv (Wood, 2006).
GAMs that are linear or can be approximated by linear functions can be substituted by simple linear regressions, thereby providing coefficients describing the sign and strength of the interactions. When applying this methodology to the male time series using a 99% confidence level, 501 out of 552 significant GAMs could be replaced by linear models (Figure 3b). Out of the 501 linear interactions roughly 75% are negative, which agrees with general expectations (Foster and Bell, 2012). Note that the matrix of interaction coefficients is not symmetric because interactions between taxa need not have the same form, or even be reciprocal. The full set of models can then form the basis for the construction of a comprehensive interaction network (Figure 3c).
Complementary approaches
The reigning tenet in ecology is that the more similar two groups of organisms are, both in terms of phylogeny and resource use, the stronger the competitive interaction (Violle et al., 2011). However, it has been demonstrated that competitive interactions can occur among widely different taxa (Freilich et al., 2010), and the dynamics of distantly related phyla can be tightly coupled (Trosvik et al., 2010). A recently developed approach, termed ‘reverse ecology’, uses this insight to infer biotic interactions (Levy and Borenstein, 2012). In this framework, the metabolic networks of species are reconstructed from their genome sequences. Negative interactions are inferred from the degree of overlap in external resource use in a species pair, whereas positive interactions are inferred from the number of internally produced metabolites in one species that can function as external resources for the other species. Although this approach is very appealing and can certainly be informative (Levy and Borenstein, 2013) it only estimates the biotic interaction potential, but it constitutes a valuable means of generating hypotheses and validating results of other approaches.
Future directions
Although the approach described here is, in some senses, simplistic it should provide an accurate representation of the interaction structure in a complex microbial community given appropriate data. The reason for using regression models with single independent variables has to do with the problem of model selection. This can be tricky enough for a single model with many covariates and for a large set of complex models automated model selection will be required. Although manual model inspection is generally preferable, there are several tools available for automated term selection (Calcagno and de Mazancourt, 2010; Marra and Wood, 2011), which should be further explored. Furthermore, alternative approaches to time series modelling, like the one recently described by Stein et al. (2013), provide a basis for comparison of modelling results.
We argue that there is no substitute for observing actual community dynamics in order to reliably infer and quantify biotic interactions between populations in the gut microbiota or other complex communities. Thus, longitudinal sampling of appropriate frequency and resolution should be carefully considered as the next generation of microbial community studies are designed.
References
Biggs R, Carpenter SR, Brock WA . (2009). Turning back from the brink: detecting an impending regime shift in time to avert it. Proc Natl Acad Sci USA 106: 826–831.
Calcagno V, de Mazancourt C . (2010). Glmulti: an R package for easy automated model selection with (generalized) linear models. J Stat Softw 34: 1–29.
Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J et al. (2011). Moving pictures of the human microbiome. Genome Biol 12: R50.
Cho I, Blaser MJ . (2012). The human microbiome: at the interface of health and disease. Nat Rev Genet 13: 260–270.
Clemente JC, Ursell LK, Parfrey LW, Knight R . (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563.
de Muinck EJ, Stenseth NC, Sachse D, vander Roost J, Ronningen KS, Rudi K et al. (2013). Context-dependent competition in a model gut bacterial community. Plos One 8: e67210.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb 72: 5069–5072.
Dethlefsen L, Huse S, Sogin ML, Relman DA . (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6: e280.
Dethlefsen L, Relman DA . (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108(Suppl 1): 4554–4561.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al. (2013). The long-term stability of the human gut microbiota. Science 341: 1237439.
Faust K, Raes J . (2012). Microbial interactions: from networks to models. Nat Rev Microbiol 10: 538–550.
Ferrari MJ, Grais RF, Bharti N, Conlan AJ, Bjornstad ON, Wolfson LJ et al. (2008). The dynamics of measles in sub-Saharan Africa. Nature 451: 679–684.
Foster KR, Bell T . (2012). Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol 22: 1845–1850.
Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E . (2010). The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res 38: 3857–3868.
Gerber GK, Onderdonk AB, Bry L . (2012). Inferring dynamic signatures of microbes in complex host ecosystems. PLoS Comput Biol 8: e1002624.
Granger CWJ . (1969). Investigating causal relations by econometric models and cross-spectral methods. Econometrica 37: 414.
Greenblum S, Chiu HC, Levy R, Carr R, Borenstein E . (2013). Towards a predictive systems-level model of the human microbiome: progress, challenges, and opportunities. Curr Opin Biotech 24: 810–820.
Hastie T, Tibshirani R . (1990) Generalized Additive Models 1st edn Chapman and Hall: London; New York.
Hooper LV, Littman DR, Macpherson AJ . (2012). Interactions between the microbiota and the immune system. Science 336: 1268–1273.
Human Microbiome Project C. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214.
Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108: 4578–4585.
Levy R, Borenstein E . (2012). Reverse ecology: from systems to environments and back. Adv Exp Med Biol 751: 329–345.
Levy R, Borenstein E . (2013). Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci USA 110: 12804–12809.
Liou AP, Paziuk M, Luevano JM Jr., Machineni S, Turnbaugh PJ, Kaplan LM . (2013). Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Trans Med 5: 178ra141.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R . (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230.
Marra G, Wood SN . (2011). Practical variable selection for generalized additive models. Comput Stat Data An 55: 2372–2387.
Mazmanian SK, Round JL, Kasper DL . (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453: 620–625.
Mizrahi-Man O, Davenport ER, Gilad Y . (2013). Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. Plos One 8: e53608.
Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA et al. (2011). Strain-resolved community genomic analysis of gut microbial colonization in a premature infant (vol 108, pg 1128, 2011). Proc Natl Acad Sci USA 108: 4512–4512.
Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO . (2007). Development of the human infant intestinal microbiota. PLoS Biol 5: e177.
R Core Team. (2014) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, URL http://www.R-project.org/.
Rajilic-Stojanovic M, Heilig HG, Tims S, Zoetendal EG, de Vos WM . (2012). Long-term monitoring of the human intestinal microbiota composition. Environ Microbiol 15: 1146–1159.
Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF . (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23: 111–120.
Soergel DAW, Dey N, Knight R, Brenner SE . (2012). Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J 6: 1440–1444.
Sommer F, Backhed F . (2013). The gut microbiota–masters of host development and physiology. Nat Rev Microbiol 11: 227–238.
Stein RR, Bucci V, Toussaint NC, Buffie CG, Ratsch G, Pamer EG et al. (2013). Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput Biol 9: e1003388.
Stenseth NC, Falck W, Bjornstad ON, Krebs CJ . (1997). Population regulation in snowshoe hare and Canadian lynx: asymmetric food web configurations between hare and lynx. Proc Natl Acad Sci USA 94: 5147–5152.
Sugihara G, May R, Ye H, Hsieh CH, Deyle E, Fogarty M et al. (2012). Detecting causality in complex ecosystems. Science 338: 496–500.
Trosvik P, Stenseth NC, Rudi K . (2010). Convergent temporal dynamics of the human infant gut microbiota. ISME J 4: 151–158.
Turchin P, Taylor AD . (1992). Complex dynamics in ecological time-series. Ecology 73: 289–305.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–U487.
van Dijk EL, Jaszczyszyn Y, Thermes C . (2014). Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res 322: 12–20.
Violle C, Nemergut DR, Pu ZC, Jiang L . (2011). Phylogenetic limiting similarity and competitive exclusion. Ecol Lett 14: 782–787.
Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG et al. (2012). Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6: 94–103.
Wood SN . (2006) Generalized Additive Models: An Introduction with R. Chapman & Hall/CRC: Boca Raton, FL.
Zoetendal EG, Collier CT, Koike S, Mackie RI, Gaskins HR . (2004). Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr 134: 465–472.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Trosvik, P., de Muinck, E. & Stenseth, N. Biotic interactions and temporal dynamics of the human gastrointestinal microbiota. ISME J 9, 533–541 (2015). https://doi.org/10.1038/ismej.2014.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.147
This article is cited by
-
Gut-host Crosstalk: Methodological and Computational Challenges
Digestive Diseases and Sciences (2020)
-
Dynamics of microbial populations mediating biogeochemical cycling in a freshwater lake
Microbiome (2018)
-
Vertebrate bacterial gut diversity: size also matters
BMC Ecology (2016)
